Мечту вызывали?
07 ноября 2016
Мечту вызывали?
- 1561
- 1
- 6
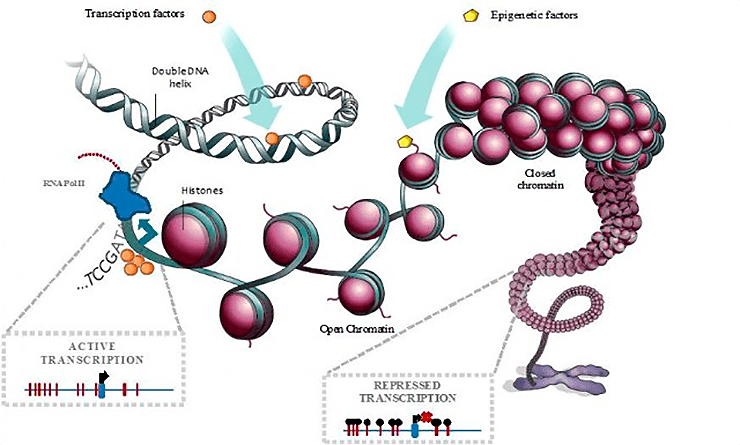
Считается, что транскрипционные факторы (ТФ) регулируют активность генов, связывая участки ДНК в открытых (доступных) районах хроматина. В ходе ENCODE-DREAM Challenge участникам соревнования предлагалось предсказать места связывания ТФ в масштабе полного генома определенного типа клеток по информации о доступности хроматина и последовательности ДНК.
сайт mappingignorance.org
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: В октябре 2016 года группа российских биоинформатиков выиграла этап престижных научных соревнований ENCODE-DREAM, приуроченный к семинару по применению методов анализа данных и машинного обучения в биологии, проходящему в рамках международной конференции ISCB-RECOMB по регуляторной и системной геномике. Предложенный российской командой алгоритм для предсказания мест связывания белков, регулирующих экспрессию генов, был признан лучшим. Однако история победы биоинформатической команды под руководством Ивана Кулаковского — это больше, чем просто «история успеха» (хотя и это дорогого стоит); эта история о том, как на наших глазах формируется и начинает работать принципиально новая модель организации науки.

«Био/мол/текст»-2016
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2016.
Генеральным спонсором конкурса, согласно нашему краудфандингу, стал предприниматель Константин Синюшин, за что ему огромный человеческий респект!
Спонсором приза зрительских симпатий выступила фирма «Атлас».
Спонсор публикации этой статьи — Алексей Петрович Семеняка.
Взрывной рост числа лабораторий и исследовательских проектов в области биологических и медицинских исследований в последние десятилетия обнажил некоторые фундаментальные проблемы в организационных принципах современной науки, которые серьезно замедляют ее развитие. В биологии этот кризис уверенно базируется на трех проблемных «китах».
Кит первый. Большие данные — большие проблемы
В «традиционной» науке дело обстояло просто: кто данные получил, тот их и проанализировал (по меньшей мере, на уровне лаборатории, хотя, как правило, даже на уровне индивидуального ученого). Ситуация изменилась, когда популярность получили высокопроизводительные методы (например, ДНК-микрочипы (microarray) [1] и секвенирование ДНК «следующего поколения» [2]), когда в результате одного эксперимента измеряется экспрессия (активность) не одного, а всех генов в образце, в десятках и сотнях образцов сразу. В биологическую науку пришли большие данные. Обработка этих гигантских числовых массивов требует владения сложными статистическими методами и специальной математической подготовки.
Логическим следствием подобной ситуации стало дальнейшее углубление специализации ученых: наряду с «мокрыми» — экспериментальными — биологическими лабораториями (wet lab) появилось множество «сухих» биоинформатических групп (dry lab) , занятых исключительно анализом данных. В идеале подобная специализация должна была многократно повысить эффективность научной работы. Однако на практике все обстоит не так радужно. Новые формы специализации упираются в традиционный формат обнародования научных данных (публикация), подразумевающий лидерство одной группы и болезненные вопросы приоритета. Кто является автором результата — группа, поставившая сложный и дорогостоящий эксперимент, или аналитики, отыскавшие в данных интересную зависимость и в конечном счете превратившие «научную информацию» в «научное знание»? Как правило, у двух сторон (биоинформатиков и биологов-экспериментаторов) ответы на этот вопрос не совпадают. В результате на свет появляются публикации, содержащие интересные, но небрежно проанализированные данные с одной стороны (экспериментаторы, не желающие «делиться»), или интересные статистические методы, не подтвержденные экспериментальной проверкой, — с другой (от биоинформатиков, не нашедших «своего» экспериментатора, готового вложиться в подтверждение или опровержение их теорий). Такое состояние дел трудно назвать оптимальным.
«Сухой» (или компьютерной) биологии на «биомолекуле» посвящено большое количество статей. Вот некоторые из них: «Вычислительное будущее биологии», «Я б в биоинформатики пошёл, пусть меня научат!», «Биоинформатика: Большие БД против „большого Р“» и «Исследовательская группа Филиппа Хайтовича, или как биологи работают с большими массивами данных» [3–6]. — Ред.
Кит второй. Кризис кооперации
Чем сильнее растет специализация в науке (как известно, настоящий специалист знает «все ни о чем»), тем очевиднее становится необходимость во встречном «синтетическом» движении — обобщении, научном сотрудничестве, общении ученых разных направлений. Это понимают даже на государственном уровне: в Европе для подачи заявки на сколько-нибудь крупный грант необходимо объединить усилия нескольких лабораторий. Однако, положа руку на сердце, подобное полупринудительное скрещивание «ужа с ежом» ради получения финансирования редко бывает по-настоящему эффективным и прорывным. Как правило, его можно охарактеризовать лишь сдержанным определением «лучше, чем ничего».
И хотя официальной позицией мировой фундаментальной науки является свободный научный поиск, в реальной академической жизни позиция «собаки на сене» — «Это моя тема...» — тоже, к сожалению, встречается весьма часто. Отсутствие общих интересов и сильная взаимная ревность и подозрительность (обусловленные всё тем же вопросом приоритета) сильно затрудняют эффективную научную коммуникацию.
Кит третий. А судьи кто?
Работающий биоинформатический метод позволяет сильно сэкономить на экспериментах и задает новые направления научного поиска, однако с ростом числа математических подходов к той или иной биологической проблеме возникает естественный вопрос — а какой же из них все-таки лучше описывает реальность и в каких условиях?
Положение биоинформатики как «сервисной науки» при чужих данных играет скверную шутку с верифицируемостью методов. Успех публикации (определяемый рейтингом журнала и числом цитирований) — это на 90% успех оригинальных данных и лишь на 10% успех аналитического метода. Биоинформатики, сотрудничающие с успешными экспериментальными лабораториями, таким образом, имеют существенное преимущество перед коллегами, практически не зависящее от качества используемых ими подходов и алгоритмов. Методы, опубликованные в Nature или Cell, — это не обязательно самые лучшие методы. Но как при таком очевидном неравенстве приоритетного доступа к данным определить лучших? Вопрос верифицируемости биоинформатических методов — это третий проблемный «кит» современной биологической науки.
Выход там же где и вход — стратегии преодоления кризиса
Альтернативой полупринудительному «грантовому» сотрудничеству является система научных сетей, активно внедряемая по всему миру энтузиастами, самым известным из которых является ученый и инноватор Стивен Френд (Steven Friend). Основанная им некоммерческая организация Sage Bionetworks занимается популяризацией «открытой науки» и развивает облачную платформу Synapse, на которой ученые разных стран могут объединяться над решением интересующей их проблемы (как правило, связанной с анализом больших данных).
Параллельно в биологии организуется всё больше международных некоммерческих консорциумов, объединяющих и экспериментаторов, и биоинформатиков, интересующихся конкретной широкой тематикой. Одни явно ориентированы на исследование конкретных заболеваний (например, консорциумы TCGA и ICGC занимаются систематическим исследованием различных типов рака). Другие больше нацелены на исследование фундаментальных вопросов (как, например, ENCODE и FANTOM, изучающие регуляцию активности генов в различных типах клеток).
Другим подходом, направленным на решение одновременно и проблемы преодоления научного изоляционизма, и проблемы верифицируемости биоинформатических методов, является система научных соревнований. Такое состязание лежит, можно сказать, у самых истоков молекулярной биологии, когда, располагая практически одними и теми же данными, Уотсон и Крик состязались с Полингом в предсказании структуры ДНК.
Первыми же массовыми состязаниями теоретиков (насколько мне известно) стал CASP — открытое и независимое состязание, которое раз в два года устраивают ученые, занимающиеся структурной биологией. Для него несколько лабораторий, занимающихся кристаллизацией и определением трехмерной структуры белков, на несколько месяцев «придерживают» свои данные, давая возможность структурным биологам состязаться в предсказании неопубликованной структуры [7]. Выигравшей считается команда теоретиков, чья модель продемонстрировала лучшее соответствие экспериментальным данным.
Аналогичный подход — проверка предсказательных методов закрытыми (до конца состязаний) экспериментальными данными — перешел и в научные состязания нового поколения, самым масштабным из которых на сегодняшний день является DREAM Challenge [8].
С момента старта в 2006 году в рамках инициативы DREAM (Dialogue for Reverse Engineering Assessment and Methods) под флагом научного краудсорсинга было предложено и успешно решено более десятка вычислительных задач из различных областей молекулярной и теоретической биологии. Платформой для организации соревнований уже несколько лет выступает облачная экосистема Synapse.
Временные рамки, заданные соревнованием, позволяют избежать «расхлябанности», обычной, увы, при неформальном сотрудничестве, а «пряником», привлекающим ученых в это состязание, является публикация результатов победителей в престижном научном журнале и приглашение с выступлением на конференцию. Таким образом, для молодых ученых эти состязания — настоящий шанс одним прыжком попасть в «большую науку». Но важнее другое — DREAM Challenge формирует новую биоинформатическую «табель о рангах», более объективную и независимую, чем традиционный «рейтинг публикаций», и постепенно разворачивает научное сообщество в направлении открытого коллективного мозгового штурма актуальных научных проблем в противовес традиционному «собственническому» подходу. На наших глазах формируется фактически независимый научный бренд, и, возможно, уже через несколько лет «протестировано и подтверждено — DREAM Challenge» будет значить не меньше, чем «опубликовано в Nature».
DREAM Challenge 2016
В 2016 году в рамках DREAM проходит несколько параллельных соревнований. Темой совместного проекта консорциумов ENCODE и DREAM было предсказание участков ДНК, связывающих факторы транскрипции на основании экспериментальной информации о доступности хроматина и компьютерного анализа геномной последовательности.
Как известно, все клетки многоклеточного организма содержат одну и ту же ДНК (геном), однако обладают разными свойствами: в организме человека, например, насчитывается несколько сотен специализированных типов клеток. В разных клетках работают (экспрессируются) разные наборы генов, за счет этого и достигается такое разнообразие тканей организма. И хотя ученые давно расшифровали генетический код [10], мы до сих пор плохо понимаем, как именно гены включаются и выключаются.
Известно, что важную роль в этом играют транскрипционные факторы — белки, которые садятся на ДНК в особых точках (сайтах связывания) и привлекают туда РНК-полимеразу, считывающую с гена мРНК, необходимую для синтеза определенного белка.
Другим, более общим, механизмом, регулирующим активность генов в клетке, является упаковка хроматина (комплекса ДНК и основных упаковочных белков-гистонов). Считается, что гены в плотно упакованной фракции хроматина выключены и недоступны действию факторов транскрипции, а в «открытом» хроматине — наоборот.
Экспериментальное определение мест связывания тех или иных белков с генами — дорогостоящая и сложная процедура, которую необходимо повторять для каждого белка-регулятора (а их у человека насчитывается до полутора тысяч) отдельно в каждом типе клеток. Определение доступности хроматина — значительно более простая операция, и для каждого типа клеток ее достаточно провести один раз. Поэтому было бы очень заманчиво создать компьютерный метод, способный воссоздать профиль связывания факторов транскрипции с геномом, основываясь на доступности хроматина и известной информации о геномной последовательности.
Из теоретических оснований вывести точную формулу, по которой можно было бы сказать, садится ли белок на этот участок ДНК или нет, — не получается: слишком многие факторы влияют на этот процесс. Однако в известных экспериментальных данных можно попытаться выявить закономерности, описывающие интересующую нас характеристику. Методы выявления зависимостей в данных — это то, что в современном мире называют машинным обучением. Типичная задача машинного обучения — по одному эксперименту понять закономерность (обучиться), чтобы потом предсказать результат другого эксперимента. Но здесь есть одна тонкость: большая часть современных молекулярно-биологических данных получена для раковых клеточных линий [11] (с ними проще работать в лаборатории), в то время как наибольший интерес для исследователей представляют нормальные клетки. Как эффективно экстраполировать раковые данные на нормальные ткани? Именно в этом и заключался вызов (challenge!) соревнований этого года.
Для ENCODE-DREAM методы «машинного обучения» отрабатывались участниками на известных экспериментальных данных по сайтам связывания транскрипционных факторов в раковых клеточных линиях, а потом проверялись организаторами на неопубликованных данных по связыванию факторов транскрипции в нормальных клетках печени.
Лучше меньше, да лучше! Секреты успеха аутосомной команды
Считается, что алгоритмы машинного обучения работают тем лучше, чем больше данных они пропустили через себя на стадии обучения. Новаторство команды autosome.ru, обеспечившее им в конечном счете первое место, состояло в том, чтобы целенаправленно ограничить набор данных и использовать для обучения алгоритма информацию лишь из похожих клеточных линий.
Вторым фактором успеха было использование мощного метода машинного обучения XGBoost, который работает как ансамбль решающих деревьев [12].
И наконец, третьим фактором стала кропотливая работа по конструированию и отбору признаков (или «фичей» — features, как их называют на языке машинного обучения), которые помогают отличать связанные участки генома от несвязанных. Некоторые признаки очень слабо подтверждают или опровергают гипотезу о связывании фактора с ДНК, но алгоритмы машинного обучения способны учитывать даже слабые закономерности и c их помощью улучшать модель. Одни признаки дают прирост качества предсказания на десятки процентов, другие — на десятые доли процента, но когда удается объединить «свидетельства» от десятков таких признаков, суммарный эффект получается значительным.
Вместе эти подходы завоевали не просто «техническую победу», но убедительное превосходство над соперниками. Итоговый «обобщенный рейтинг» команды autosome.ru (рис. 2), построенный по различным оценкам качества предсказаний для 12 факторов транскрипции, получился почти в два раза выше, чем у ближайшего конкурента — команды J-Team, занявшей второе место. В ближайшем будущем результаты этого первого этапа DREAM challenge будут доложены на конференции.

Рисунок 2. Знать своих героев! Слева направо члены российской (преимущественно) команды autosome.ru: Илья Воронцов, аспирант Института общей генетики имени Н.И. Вавилова РАН (Москва, Россия). Андрей Ландо, магистрант Московского физико-технического института (Долгопрудный, Россия). Григорий Сапунов, сооснователь Intento. Чебурашка, маскот. Руководитель команды — Иван Кулаковский, ведущий научный сотрудник Института молекулярной биологии имени В.А. Энгельгардта РАН (Москва, Россия). См. список публикаций Ивана. Ирина Елисеева, научный сотрудник Института белка РАН (Пущино, Россия). Валентина Боева, зав. лабораторией Института Кошан (Париж, Франция), окончила мехмат МГУ и защитила кандидатскую диссертацию в России. Всеволод Макеев, член-корр. РАН, зав. лабораторией Института общей генетики имени Вавилова РАН (Москва, Россия). См. список публикаций Всеволода.
До недавнего времени биоинформатики, вполне осознавая свое зависимое (от экспериментаторов — производителей данных) положение, грустно шутили, что, несмотря на головокружительные успехи их науки, ожидать присуждение Нобелевской премии за анализ данных в ближайшем будущем не приходится . Однако соревнования, подобные DREAM Challenge, меняют привычное (и несправедливое) положение вещей, и, возможно, нам все-таки доведется увидеть автора какого-нибудь сногсшибательного алгоритма на приеме у шведского короля. Но даже если нет, то, разворачивая науку от самолюбивой «борьбы за приоритет» в направлении коллективного поиска истины, основанного на сотрудничестве (на заключительном — краудсорсинговом — этапе все команды, принимающие участие в состязании, обменяются данными и алгоритмами и будут вместе обсуждать выигрышные стратегии), такие состязания выступают своеобразным «прототипом» науки будущего, более открытой, свободной и эффективной, чем наука сегодняшнего дня, готовой к новым вызовам (challenges!) и новым мечтам (dreams!).
Теперь биоинформатикам приходится искать новый предмет для шуток — в 2013 году Нобелевскую премию за разработку методов моделирования больших и сложных химических систем и реакций получили Мартин Карплюс, Майкл Левитт и Ариэ Варшель: «„Виртуальная“ Нобелевская премия по химии (2013)» [13]. — Ред.
Неформальная команда autosome.ru объединяет исследователей из нескольких институтов (в первую очередь ИМБ РАН и ИОГен РАН), работающих в области регуляторной геномики и биоинформатики. Участниками в разном составе в разные годы была разработана масса вычислительных методов и баз данных по анализу генной регуляции у эукариот (примеры работ можно найти на сайте). Среди прошлых успешных проектов можно отметить работу российской группы под руководством Всеволода Макеева в международном консорциуме FANTOM5. Участие в DREAM — один из многих совместных проектов, в ходе которого активно использовались предыдущие наработки.
Автор выражает благодарность членам команды autosome.ru за помощь в работе над статьей и Андрею Зиновьеву (Институт Кюри, Париж) за интересную дискуссию о путях развития науки о данных.
Литература
- Важнейшие методы молекулярной биологии и генной инженерии;
- 454-секвенирование (высокопроизводительное пиросеквенирование ДНК);
- Вычислительное будущее биологии;
- Я б в биоинформатики пошёл, пусть меня научат!;
- Биоинформатика: большие БД против «большого Р»;
- Исследовательская группа Филиппа Хайтовича, или Как биологи работают с большими массивами данных;
- Торжество компьютерных методов: предсказание строения белков;
- Bender E. (2016). Challenges: crowdsourced solutions. Nature. 533, S62–S64;
- Новый метод CETCh-seq может за одну метку поймать много результатов;
- У истоков генетического кода: родственные души;
- Бессмертные клетки Генриетты Лакс;
- Chen T. and Guestrin C. (2016). XGBoost: reliable large-scale tree boosting system. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 785–794;
- «Виртуальная» Нобелевская премия по химии (2013).

Комментарии


