Важнейшие методы молекулярной биологии и генной инженерии
27 октября 2011
Важнейшие методы молекулярной биологии и генной инженерии
- 70396
- 10
- 78

Молекулярно-биологическая лаборатория.
Университет Ватерлоо (Канада).
-
Автор
-
Редакторы
Темы
Статья на конкурс «био/мол/текст»: Биология — самая быстро развивающаяся наука во второй половине ХХ и ХХI веке. Связано это, в первую очередь, с появлением нового ее раздела — молекулярной биологии, подоплекой возникновения которой, в свою очередь, стало стремительное развитие физики, химии и физико-химических методов. Я расскажу о важнейших (на мой взгляд) методах молекулярной биологии, с помощью которых были сделаны многие открытия, известные не только в узких научных кругах, но и среди широкой публики. Они принесли множество Нобелевских премий как тем, кто их открыл, так и тем, кто их использовал. Многие из них применяются не только в биологии, но и в других областях: медицине, криминалистике, археологии.

«Био/мол/текст»-2011
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2011 в номинации «Лучшая обзорная статья».
Введение
Строение ДНК
Началом молекулярной биологии принято считать открытие структуры ДНК (рис. 1) в 1953 году Джеймсом Уотсоном и Френсисом Криком, за что они (совместно с Морисом Уилкинсом) в 1962 году получили Нобелевскую премию по физиологии и медицине [1], [2]. Они выяснили, что молекула ДНК представляет из себя две противоположно направленные цепочки полинуклеотидов, закрученных вокруг общей оси в двойную спираль, причем друг напротив друга в спирали всегда стоят определенные азотистые основания: напротив гуанина (Г или G) — цитозин (Ц или C), а напротив аденина (А) — тимин (Т) (рис. 1). Это называют правилом комплементарости: цепи удерживаются вместе за счет водородных связей, возникающих между нуклеотидами. Водородная связь гораздо слабее ковалентной, с помощью которой нуклеотидные остатки соединяются между собой в одной цепи ДНК, формируя так называемый сахаро-фосфатный остов. Его так называют, поскольку в нем остатки сахара (дезоксирибозы) в нуклеотидах связаны друг с другом через остатки ортофосфорной кислоты — фосфаты. Концы обеих цепей не равноценны: по порядковому номеру атома углерода в остатке сахара один из них называют 3´, а другой — 5´. Синтез ДНК (как и РНК) в природе, как правило, идет от 5´ к 3´-концу.
Возможно, следовало бы начать отсчет с экспериментов Бидла, Татума, Ледерберга, но это дело вкуса. — Ред.

Рисунок 1. Схема строения двуцепочечной молекулы ДНК.
Однако ДНК не обязательно бывает двуцепочечной — иногда встречаются и одноцепочечные молекулы (например, в геномах некоторых вирусов). Это очень важно, поскольку, как будет рассказано ниже, двуцепочечные молекулы могут денатурировать на одноцепочечные, и, наоборот, одноцепочечные образовывать двуцепочечные.
Строение РНК аналогично (хотя обычно она состоит из одной цепи и часто образует комплементарные взаимодействия между участками одной молекулы), только вместо тимина в ее состав входит урацил, а вместо дезоксирибозы — рибоза. Подробнее обо всем этом написано в учебниках по молекулярной биологии [3].
Центральная догма молекулярной биологии
Я кратко напомню так называемую центральную догму молекулярной биологии, в первоначальном виде сформулированную Фрэнсисом Криком [4]. В общем случае она гласит, что генетическая информация при реализации передается от нуклеиновых кислот к белку, но не наоборот. А точнее, возможно передача ДНК → ДНК (репликация), ДНК → РНК (транскрипция) и РНК → белок (трансляция). Так же существуют значительно реже реализуемые пути, свойственные некоторым вирусам: РНК → ДНК (обратная транскрипция) и РНК → РНК (репликация РНК). Также напомню, что белки состоят из аминокислотных остатков, последовательность которых закодирована в генетическом коде организма: три нуклеотида (их называют кодон, или триплет) кодируют одну аминокислоту, причем одну и ту же аминокислоту может кодировать несколько кодонов.
Во второй половине XX века получили развитие технологии рекомбинантной ДНК (то есть, методы манипуляции ДНК, позволяющие различными способами изменять последовательность и состав нуклеотидов в молекуле). Именно на их основе происходит развитие всех молекулярно-биологических методов и поныне, хотя они стали значительно сложнее, как идейно, так и технологически. Именно молекулярная биология вызвала такой бурный рост количества биологической информации за последние полвека.
Я расскажу о методах манипуляции и изучения ДНК и РНК, совсем немного коснусь белков, поскольку в основном методы, связанные с ними, ближе к биохимии, чем к молекулярной биологии (хотя грань между ними в последнее время стала очень расплывчатой).
Разрезание и сшивание
Рестрикционные эндонуклеазы

Рисунок 2. Сайты рестрикции. Сверху — целевая последовательность рестриктазы SmaI, при работе которой образуются «тупые» концы. Снизу — целевая последовательность рестриктазы EcoRI, при работе которой образуются «липкие» концы.
Одним из первых и важнейших из шагов молекулярной биологии стала возможность разрезать молекулы ДНК, причем в строго определенных местах [3]. Этот метод был изобретен при изучении в 1950—1970-е годы такого феномена: некоторые виды бактерий при добавлении в среду чужеродной ДНК разрушали ее, в то время, как их собственная ДНК оставалась невредимой. Оказалось, что они для этого используют ферменты, позднее названные рестрикционными нуклеазами или рестриктазами. Существует множество видов рестриктаз: к 2007-му году их было известно более 3000 [5]. Важным свойством каждого подобного фермента является его способность разрезать строго определенную — целевую — последовательность нуклеотидов ДНК (рис. 2). Рестриктазы не воздействуют на собственную ДНК клетки, поскольку нуклеотиды в целевых последовательностях модифицированы так, что рестриктаза не может с ними работать. (Правда, иногда, наоборот, они могут разрезать только модифицированные последовательности — для борьбы с теми, кто модифицирует ДНК, защищаясь от вышеописанных рестриктаз.) Из-за того, что целевые последовательности бывают различной длины, частота встречаемости их в молекулах ДНК варьирует: чем длиннее необходимый фрагмент, тем меньше вероятность его появления. Соответственно, образующиеся при обработке различными рестриктазами фрагменты ДНК будут иметь различную длину.
Новые эндонуклеазы продолжают открывать и по сей день. Многие из них до сих пор не клонированы, то есть, не известны гены, которые их кодируют, и в качестве «фермента» используют некую очищенную фракцию белков, обладающую нужной каталитической активностью. Новосибирская компания СибЭнзим долгое время успешно соревновалась с компанией New England Biolabs — признанным во всем мире лидером по поставке рестритаз (то есть предлагала такое же или большее различных рестриктаз, некоторые из которых весьма экзотичны). — Ред.
За выделение первой рестриктазы, изучение ее свойств и первое применение для картирования хромосом Вернер Арбер (Werner Arber), Дэн Натанс (Dan Nathans) и Гамильтон Смит (Hamilton Smith) в 1978 году получили Нобелевскую премию по физиологии и медицине.
ДНК-лигазы
Для создания новых молекул ДНК, разумеется, кроме разрезания, необходима еще и возможность сшивания двух цепей. Это делают с помощью ферментов, называемых ДНК-лигазами, которые сшивают сахаро-фосфатный остов двух цепей ДНК. Поскольку по химическому строению ДНК не отличается у разных организмов, можно сшивать ДНК из любых источников, и клетка не сможет отличить полученную молекулу от своей собственной ДНК.
Разделение молекул ДНК: электрофорез в геле
Часто приходится иметь дело со смесью молекул ДНК разной длины. Например, при обработке химически выделенной из организма ДНК рестриктазами как раз получится смесь фрагментов ДНК, причем их длины будут различаться.
Поскольку любая молекула ДНК в водном растворе отрицательно заряжена, появляется возможность разделить смесь фрагментов ДНК различных размеров по их длине с помощью электрофореза [3], [6]. ДНК помещают в гель (обычно, агарозный для относительно длинных и сильно отличающихся молекул или полиакриламидный для электрофореза с высоким разрешением), который помещают в постоянное электрическое поле. Из-за этого молекулы ДНК будут двигаться к положительному электроду (аноду), причем их скорости будут зависеть от длины молекулы: чем она длиннее, тем сильнее ей мешает двигаться гель и, соответственно, тем ниже скорость. После электрофореза смеси фрагментов разных длин в геле образуют полосы, соответствующие фрагментам одной и той же длины. С помощью маркеров (смесей фрагментов ДНК известных длин) можно установить длину молекул в образце (рис. 3).

Рисунок 3. Схема проведения электрофореза ДНК в агарозном геле.
сайт Molecular Station
Визуализовать результаты фореза можно двумя способами. Первый, наиболее часто используемый в последнее время — добавление в гель веществ, флуоресцирующих в присутствии ДНК (традиционно использовался довольно токсичный бромистый этидий; в последнее время в обиход входят более безопасные вещества). Бромистый этидий светится оранжевым светом при облучении ультрафиолетом, причем при связывании с ДНК интенсивность свечения возрастает на несколько порядков (рис. 4). Другой метод заключается в использовании радиоактивных изотопов, которые необходимо предварительно включить в состав анализируемой ДНК. В этом случае на гель сверху кладут фотопластинку, которая засвечивается над полосами ДНК за счет радиоактивного излучения (этот метод визуализации называют авторадиографией).
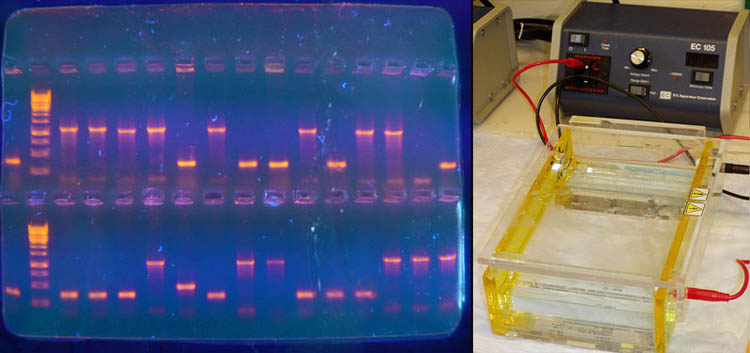
Рисунок 4. Электрофорез в агарозном геле с использованием бромистого этидия для визуализации результатов в ультрафиолете (слева). Вторая слева дорожка — маркер с известными длинами фрагментов. Справа — Установка для проведения электрофореза в геле.
Кроме «обычного» электрофореза в пластине из геля, в некоторых случаях используют капиллярный электрофорез, который проводят в очень тонкой трубочке, наполненной гелем (обычно полиакриламидным). Разрешающая способность такого электрофореза значительно выше: с его помощью можно разделять молекулы ДНК, отличающиеся по длине всего на один нуклеотид. Об одном из важных приложений такого метода читайте ниже в описании метода секвенирования ДНК по Сэнгеру.
Выявление определенной последовательности ДНК в смеси. Саузерн блоттинг
С помощью электрофореза можно узнать размер молекул ДНК в растворе, однако он ничего не скажет о последовательности нуклеотидов в них. С помощью гибридизации ДНК можно понять, какая из полос содержит фрагмент со строго определенной последовательностью. Гибридизация ДНК основана на образовании водородных связей между двумя цепями ДНК, приводящем к их соединению [3], [7].
Сначала необходимо синтезировать ДНК-зонд, комплементарный той последовательности, которую мы ищем. Он обычно представляет собой одноцепочечную молекулу ДНК длиной 10–1000 нуклеотидов. Из-за комплементарности зонд свяжется с необходимой последовательностью, а за счет флуоресцентной метки или радиоизотопов, встроенных в зонд, результаты можно увидеть.
Для этого используют процедуру, называемую Саузерн-блоттинг или перенос по Саузерну, названную по имени ученого, ее изобретшего (Edwin Southern). Первоначально смесь фрагментов ДНК разделяют с помощью электрофореза. На гель сверху кладут лист нитроцеллюлозы или нейлона, и разделенные фрагменты ДНК переносятся на него за счет блоттинга: гель лежит на губке в ванночке с раствором щелочи, который просачивается через гель и нитроцеллюлозу за счет капиллярного эффекта от бумажных полотенец, сложенных сверху. Во время просачивания щелочь вызывает денатурацию ДНК, и на поверхность пластины нитроцеллюлозы переносятся и закрепляются там уже одноцепочечные фрагменты. Лист нитроцеллюлозы аккуратно снимают с геля и обрабатывают радиоактивно меченной ДНК-пробой, специфичной к необходимой последовательности ДНК. Лист нитроцеллюлозы тщательно отмывают, чтобы на нем остались только те молекулы пробы, которые гибридизовались с ДНК на нитроцеллюлозе. После авторадиографии ДНК, с которой гибридизовался зонд, будет видна как полосы на фотопластинке (рис. 5).
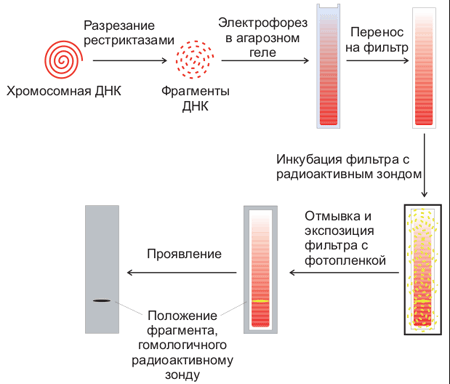
Рисунок 5. Схема проведения Саузерн-блоттинга.
Адаптация этой методики для определения специфических последовательностей РНК называется, в противоположность Саузерн-блоттингу, норзерн-блоттингом (northern blotting: southern по-английски означает «южный», а northern — «северный»). В этом случае проводят электрофорез в геле с молекулами мРНК, а в качестве зонда выбирают одноцепочечную молекулу ДНК или РНК.
Клонирование ДНК
Мы уже знаем, каким образом можно разрезать геном на части (а их сшивать с произвольными молекулами ДНК), разделять полученные фрагменты по длине и с помощью гибридизации выбрать необходимый. Теперь настало время узнать, как, скомбинировав эти методы, мы можем клонировать участок генома (например, определенный ген). В геноме любой ген занимает крайне маленькую длину (по сравнению со всей ДНК клетки). Клонирование ДНК буквально означает создание большого числа копий определенного ее фрагмента. Именно за счет этой амплификации мы получаем возможность выделить участок ДНК и получить его в достаточном для изучения количестве.
Каким образом разделить фрагменты ДНК по длине и идентифицировать нужный — было рассказано выше. Теперь надо понять, каким образом можно копировать необходимый нам фрагмент. Существует два основных метода: использование быстро делящихся организмов (обычно бактерий Escherichia coli — кишечной палочки — или дрожжей Saccharomyces serevisiae) или проделать аналогичный процесс, но in vitro с помощью полимеразной цепной реакции.
Репликация в бактериях
Поскольку при каждом клеточном делении бактерии (как и любые другие клетки, не считая предшественников половых клеток) удваивают свою ДНК, это можно использовать для умножения количества необходимой нам ДНК [3]. Для того, чтобы внедрить наш фрагмент ДНК в бактерию, необходимо «вшить» его в специальный вектор, в качестве которого обычно используют бактериальную плазмиду (небольшую — относительно бактериальной хромосомы — кольцевую молекулу ДНК, реплицирующуюся отдельно от хромосомы). У бактерий «дикого типа» часто встречаются подобные структуры: они часто переносятся «горизонтально» между разными штаммами или даже видами бактерий. Чаще всего в них содержатся гены устойчивости к антибиотикам (именно из-за этого свойства их и открыли) или бактериофагам, а также гены, позволяющие клетке использовать более разнообразный субстрат. (Иногда же они «эгоистичны» и не несут никаких функций.) Именно такие плазмиды обычно и используют в молекулярно-генетических исследованиях. В плазмидах обязательно содержится точка начала репликации (последовательность, с которой начинается репликация молекулы), целевая последовательность рестриктазы и ген, позволяющий отобрать те клетки, которые обладают этой плазмидой (обычно, это гены устойчивости к какому-нибудь антибиотику). В некоторых случаях (например, при изучении очень больших фрагментов ДНК) используют не плазмиду, а искусственную бактериальную хромосому.
В плазмиду с помощью рестриктаз и лигаз встраивают необходимый фрагмент ДНК, после чего добавляют ее в культуру бактерий при специальных условиях, обеспечивающих трансформацию — процесс активного захвата бактерией ДНК из внешней среды (рис. 6). После этого проводят отбор бактерий, трансформация которых прошла успешно, добавляя соответствующий гену в плазмиде антибиотик: в живых остаются только клетки, несущие ген устойчивости (а, следовательно, и плазмиду). Далее, после роста культуры клеток, из нее выделяют плазмиды, а из них с помощью рестриктаз выделяют «наш» фрагмент ДНК (или использую плазмиду целиком). Если же ген вставили в плазмиду для того, чтобы получить его белковый продукт, необходимо обеспечить культуре условия для роста, а потом просто выделить требуемый белок.
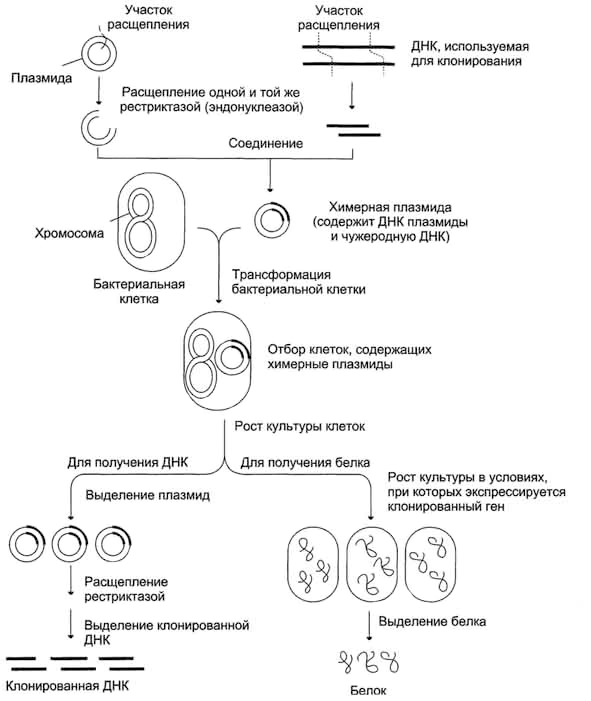
Рисунок 6. Схема клонирования участка ДНК (гена) в бактериях.
сайт biochemistry.ru
На этом месте сразу же должен возникать вопрос: как же все это возможно было использовать до того, когда были расшифрованы геномы, да и чтение последовательности ДНК было еще дорогим и малораспространенным? Положим, с помощью рестрикции и клонирования полученных фрагментов мы получим библиотеку ДНК, то есть набор бактерий, несущих различные плазмиды, содержащие суммарно весь геном (или заметную его часть). Но каким образом мы сможем понять, в каком из фрагментов содержится необходимый ген? Для этого использовали метод гибридизации. Сначала необходимо было выделить белок нужного гена. После чего отсеквенировать его фрагмент, обратить генетический код и получить последовательность нуклеотидов (конечно, из-за вырожденности генетического кода приходилось пробовать много различных вариантов). В соответствии с ней химически синтезировали короткую молекулу ДНК, которую и использовали в качестве зонда для гибридизации.
Но в некоторых случаях этот метод давал сбои — например, так произошло с фактором свертывания крови VIII. Этот белок участвует в свертывании крови , и нарушения в его функциональности являются причиной одного из самых распространенных генетических заболеваний — гемофилии А. Раньше для лечения приходилось выделять этот белок из большого числа организмов, потому что не удавалось клонировать его для производства бактериями. Связано это было с тем, что его длина составляет около 180000 пар нуклеотидов, и он содержит много интронов (некодирующих фрагментов между кодирующими) — неудивительно, что ни в одну плазмиду этот ген не попал целиком.
О механизмах свертывания крови см. «Как работает свертывание крови?». — Ред.
Полимеразная цепная реакция (ПЦР)
Полимеразная цепная реакция — молекулярно-биологический метод, позволяющий добиться колоссального (до 1012 раз) увеличения числа копий определенного фрагмента ДНК in vitro [3], [9]. Она была изобретена Кэри Муллисом (Kary Mullis) в 1983 году, за что в 1993 году он получил Нобелевскую премию по химии (совместно с М. Смитом). (См. также: «Кари Маллис, изобретатель ПЦР» [10].)
Метод основан на многократном избирательном копировании определенного участка ДНК при помощи ферментов в искусственных условиях. При этом происходит копирование только того участка ДНК, который удовлетворяет заданным условиям, и только в том случае, если он присутствует в исследуемом образце. В отличие от репликации ДНК в клетках живых организмов, с помощью ПЦР амплифицируют сравнительно короткие участки ДНК (обычно, не более 3000 пар нуклеотидов, однако есть методы позволяющие «поднимать» до 20 тысяч пар нуклеотидов — так называемый Long Range PCR).
Фактически, ПЦР является искусственной многократной репликацией фрагмента ДНК (рис. 7). ДНК-полимеразы так устроены, что не могут синтезировать новую ДНК, просто имея в наличии матрицу и мономеры. Для этого необходима еще и затравка (праймер), с которого они начинают синтез. Праймер — это короткий одноцепочечный фрагмент нуклеиновой кислоты, комплементарный ДНК-матрице. При репликации в клетке такие праймеры синтезируются специальным ферментом праймазой и являются молекулами РНК, которые позже заменяются на ДНК. Однако в ПЦР используют искусственно синтезированные молекулы ДНК, поскольку в этом случае не нужна стадия удаления РНК и синтеза на их месте ДНК. В ПЦР праймеры ограничивают амплифицируемый участок с обеих сторон.
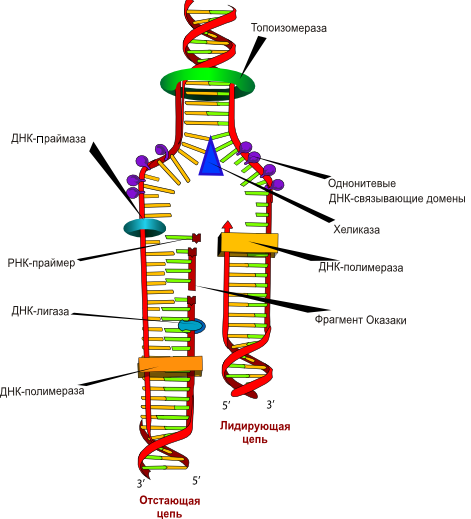
Рисунок 7. Репликация ДНК — важнейший для живых организмов процесс, основа множества молекулярно-биологических методов. Поскольку каждая из цепей ДНК содержит последовательность нуклеотидов, комплементарную другой цепи (их информационное содержание одинаково), при удвоении ДНК цепи расходятся, а затем каждая цепь служит матрицей, на которой выстраивается комплементарная ей новая цепь ДНК. В результате образуются два дуплекса ДНК, каждый из которых является точной (без учета ошибок синтеза) копией первоначальной молекулы.
Итак, пора объяснить, как же ПЦР работает. Изначально в реакционной смеси находятся: ДНК-матрица, праймеры, ДНК-полимераза, свободные нуклеозиды (будущие «буквы» в новосинтезированной ДНК), а также некоторые другие вещества, улучшающие работу полимеразы (их добавляют в специальные буферы, используемые в реакции).
Чтобы синтезировать ДНК, комплементарную матрице, необходимо, чтобы один из праймеров образовал с ней водородные связи (как говорят, «отжегся» на ней). Но ведь матрица уже образует их со второй цепью! Значит, сначала необходимо расплавить ДНК, — то есть разрушить водородные связи. Делают это с помощью простого нагревания (до ≈95 °С) — стадия, называемая денатурацией. Но теперь и праймеры из-за высокой температуры не могут отжечься на матрице! Тогда температуру понижают (50–65 °С), праймеры отжигаются, после чего температуру немного поднимают (до оптимума работы полимеразы, обычно, около 72 °С). И тогда полимераза начинает синтезировать комплементарные матрице цепи ДНК — это называют элонгацией (рис . 8). После одного такого цикла количество копий необходимых фрагментов удвоилось. Однако ничто не мешает повторить это еще раз. И не один, а несколько десятков раз! И с каждым повтором количество копий нашего фрагмента ДНК будет удваиваться, ведь новосинтезированные молекулы тоже будут служить матрицами (рис. 9)! (На самом деле эффективность ПЦР редко настолько высока, что количество копий именно удваивается, но в идеале это так, да и реальные числа часто бывают близки к этому.)
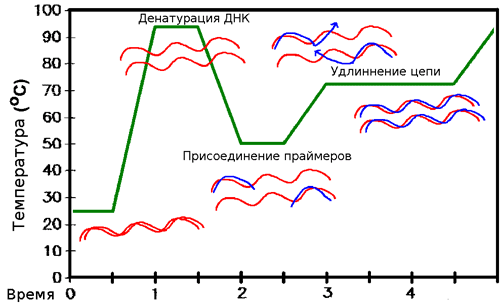
Рисунок 8. Схема ПЦР.
рисунок М. Карра
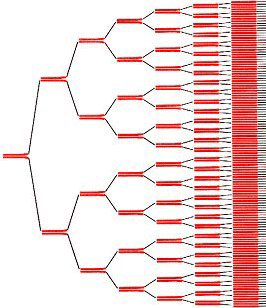
Рисунок 9. С каждым циклом ПЦР количество целевой ДНК удваивается.
Увидеть результаты ПЦР очень просто: достаточно провести электрофорез реакционной смеси после ПЦР, и будет видна яркая полоса с полученными копиями.
Раньше полимеразу, инактивирующуюся при нагревании с каждым циклом, приходилось все время добавлять, но вскоре было предложено использовать термостабильную полимеразу из термофильных бактерий, которая выдерживает такой нагрев, что сильно упростило проведение ПЦР (чаще всего используют Taq-полимеразу из бактерии Thermus aquaticus [11]).
Чтобы избежать сильного испарения воды из реакционной смеси, в нее добавляют масло, покрывающее ее сверху, и/или используют нагревающуюся крышку термоциклера — прибора, в котором проводят ПЦР. Он быстро меняет температуру пробирок, и их не приходится постоянно перекладывать из одного термостата в другой. Для предотвращения неспецифического синтеза еще до нагрева и собственно начала циклов, часто использую ПЦР с «горячим стартом»: вся ДНК и полимераза разделяются между собой парафиновой прослойкой, которая плавится при высокой температуре и дает им взаимодействовать уже в правильных условиях. Иногда же используют модифицированные полимеразы, которые не работают при низкой температуре.
Можно еще много говорить о различных тонкостях ПЦР, но важнее всего сказать об альтернативных классическому форезу методах определения результатов. Например, довольно очевидным вариантом является добавление в реакционную пробирку перед началом реакции веществ, флуоресцирующих в присутствии ДНК. Тогда, сравнив изначальную флуоресценцию с конечной, можно увидеть, синтезировалось ли значительное количество ДНК или нет. Но этот способ не специфичен: мы никак не сможем определить, синтезировался ли необходимый фрагмент, или это какие-то праймеры слиплись и достроились до непредсказуемых последовательностей.
Наиболее интересным вариантом является ПЦР «в реальном времени» («real-time PCR») . Существует несколько реализаций этого метода, но идея везде одна и та же: можно прямо в ходе реакции наблюдать за накоплением продуктов ПЦР (по флуоресценции). Соответственно, для проведения ПЦР «в реальном времени» нужен специальный прибор, способный возбуждать и считывать флуоресценцию в каждой пробирке. Самое простое решение — добавить в пробирку те же самые вещества, которые флуоресцируют в присутствии ДНК, однако минусы такого метода уже были описаны выше.
Строго это называется «ПЦР с регистрацией флуоресценции в режиме реального времени» или «количественная ПЦР». — Ред.
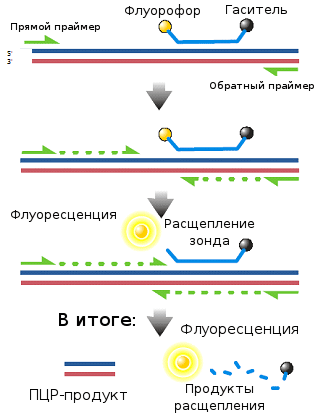
Рисунок 10. Схема работы ПЦР «в реальном времени»: Taq Man Assay.

Рисунок 11. Пример кривых накопления флуоресценции в ПЦР «в реальном времени»: зависимость интенсивности флуоресценции (в нескольких пробирках — на каждую своя кривая) от номера цикла.
сайт realtimepcr.dk
Самой популярной реализацией такого подхода является метод выщепления флуорофора за счет разрушения зонда (TaqMan Assay; рис. 10). В этом случае в реакционной смеси должен присутствовать еще один компонент — специальный одноцепочечный ДНК-зонд: молекула ДНК, комплементарная последовательности амплифицируемого фрагмента, расположенной между праймерами. При этом к одному его концу должен быть химически приделан флуорофор (флуоресцирующая молекула), а к другому — гаситель (молекула, поглощающая энергию флуорофора и «гасящая» флуоресценцию). Когда такой зонд находится в растворе или комплементарно связан с целевой последовательностью, флуорофор и гаситель находятся относительно недалеко друг от друга, и флуоресценции не наблюдается. Однако за счет 3´-экзонуклеазной активности, которой обладает Taq-полимераза (то есть она расщепляет ДНК, на которую «натыкается» в ходе синтеза, и на ее месте синтезирует новую), зонд при синтезе второй цепи разрушается, флуорофор и гаситель за счет диффузии удаляются друг от друга, и появляется флуоресценция.
Поскольку число копий в ходе ПЦР растет экспоненциально, так же растет и флуоресценция. Однако это продолжается недолго, поскольку в какой-то момент эффективность реакции начинает падать из-за постепенной инактивации полимеразы, нехватки каких-то компонентов и т. п. (рис. 11). Анализируя графики роста флуоресценции, можно много понять о протекании ПЦР, но, самое важное, можно узнать, сколько ДНК-матриц было изначально: это так называемая количественная ПЦР (quantitative PCR, qPCR).
Все варианты применения ПЦР в науке невозможно перечислить. Выделение фрагмента ДНК, секвенирование, мутагенез... ПЦР — один из самых востребованных для ненаучных целей метод (видео 1). Он широко применяется в медицине для ранней диагностики наследственных и инфекционных заболеваний, определения отцовства, в расследованиях для установления личности и для многого другого.
Видео 1. Восторг ученых по поводу изобретения ПЦР хорошо передает песня «Scientists for Better PCR» (хотя это и реклама фирмы BioRad, производящей, в том числе, оборудование и реагенты для ПЦР).
Естественные клеточные процессы in vitro
Все основные молекулярно-биологические процессы могут быть легко проведены in vitro (то есть, в пробирке). Пример приведен выше: ПЦР — это аналог репликации ДНК. Для этого достаточно просто смешать необходимые реагенты в подходящих условиях: для транскрипции нужны ДНК-матрица, РНК-полимераза и рибонуклеотиды, для трансляции — мРНК, субъединицы рибосом и аминокислоты, для обратной транскрипции — РНК-матрица, обратная транскриптаза ( она же ревертаза) и дезоксирибонуклеотиды. Эти методы широко применяются в различных областях биологии, когда необходимо, например, получить чистую РНК определенного гена. В этом случае нужно сначала провести обратную транскрипцию его (гена) мРНК, с помощью ПЦР амплифицировать ее, а затем с помощью in vitro-транскрипции получить много мРНК. Первая стадия необходима из-за того, что перед образованием зрелой мРНК в клетке проходит сплайсинг и процессинг РНК (у эукариот; у бактерий в этом смысле все проще) — подготовка к работе матрицей для синтеза белка. Иногда этого удается избежать, если вся кодирующая последовательность гена расположена в одном экзоне.
Секвенирование ДНК
Можно сказать, важнейшие методы манипуляции с ДНК уже описаны. Следующий этап — определение собственно нуклеотидной последовательности цепи в молекуле — секвенирование. Определение нуклеотидной последовательности ДНК крайне важно для множества фундаментальных и прикладных задач. Особое место оно занимает в науке: для анализа результатов секвенирования геномов была, фактически, создана новая наука — биоинформатика. Секвенированием сейчас пользуются молекулярные биологи, генетики, биохимики, микробиологи, ботаники и зоологи, и, конечно же, эволюционисты: практически вся современная систематика основана на его результатах. Секвенирование широко применяется в медицине как метод поиска наследственных заболеваний и изучения инфекций. (См., например, «Уточнение „родословной“ членистоногих» и «Скверный анекдот: негр, китаец и Крейг Вентер...». — Ред.)
На самом деле хронологически методы изобретались совсем в другом порядке. Например, секвенирование по Сэнгеру было разработано в 1977 году, а ПЦР, как говорилось выше, только в 1983-м.
Существует множество различных методик секвенирования, но все методы можно разделить на две категории: «классические» и нового поколения. Сейчас используется фактически только один «классический» метод — секвенирование по Сэнгеру , или метод терминаторов. По сравнению с новыми методами, у него есть важное преимущество: длина прочтения, то есть количество нуклеотидов в последовательности, которое можно получить за один раз, у него выше — до 1000 нуклеотидов [12]. В то же время у самого «хорошего» в этом плане «нового» метода секвенирования — 454-, или пиросеквенирования [13] — этот параметр не превышает 500 нуклеотидов . Именно длина прочтения ограничивает возможности новых методов: оказывается крайне сложно «собрать» целый геном из фрагментов размером в несколько десятков нуклеотидов. Как минимум, для этого требуются суперкомпьютеры, а некоторые места в геноме разрешить оказывается просто невозможно, если они содержат высокоповторяющиеся последовательности. В таком случае может помочь сравнение полученных фрагментов с уже имеющимся целым геномом, но таким образом невозможно прочесть геном организма впервые (de novo). (См. также: «Код жизни: прочесть не значит понять». — Ред.)
Английский биохимик и корифей молекулярной биологии, дважды лауреат Нобелевской премии по химии: за определение аминокислотной последовательности инсулина (1955 г.) и за разработку метода секвенирования ДНК (1980 г.). — Ред.
Есть метод нового поколения, позволяющий читать несколько тысяч пн, но с большими ошибками (Pacific Biosciences). 454/Roche сегодня могут читать и больше 500 пн; то же самое уже может и молодое «полупроводниковое секвенирование». — Ред.
Оба упомянутых выше метода секвенирования уже достаточно подробно описаны на «биомолекуле» [13]: очень советую ознакомиться. Я же для примера расскажу про другой распространенный быстрый и дешевый метод (в расчете на один прочитанный нуклеотид) — метод, реализованный в секвенаторах Illumina (видео 2). Основной его недостаток — чтение фрагментов очень короткой длины, не больше 100 нуклеотидов, и вытекающая отсюда сложность прочтения геном «с нуля» [14].
В этом методе можно выделить три стадии: подготовку библиотеки фрагментов (1), создание кластеров (2) и собственно секвенирование (3).
Видео 2. В интернете есть несколько хороших видео, на которых описан процесс секвенирования Illumina, например на официальном сайте компании (вкладка Technology). Правда, они все на английском языке.
- Сначала создается библиотека фрагментов ДНК из секвенируемого генома (или любого другого источника ДНК). ДНК с помощью ультразвука или специального фермента расщепляется на произвольные фрагменты длиной в несколько сотен нуклеотидов, из которых выбираются обладающие заданной длиной (выбирается экспериментатором). После этого к ним с двух концов ковалентно присоединяются различные адаптерные последовательности (рис. 12: 1);
- Этот раствор поступает на специальный микрочип с «пришитыми» к нему фрагментами ДНК, комплементарными адаптерным последовательностям, — праймерами. Там участки геномной ДНК прикрепляются с помощью адаптерных последовательностей к молекулам ДНК на чипе. С помощью ДНК-полимеразы у всех таких фрагментов достраивается вторая комплементарная цепь, которая будет уже ковалентно связана с праймером. Она случайно изгибается, и в какой-то момент попадет на праймер, комплементарный ее адаптерной последовательности. Они свяжутся, и снова достроится вторая цепь. Это повторяют несколько раз, после чего один из типов праймеров отрезают от молекулы ДНК, чтобы в образовавшемся кластере остались только одинаковые цепи (рис. 12: 2–6). Всего на чипе образуются сотни миллионов таких кластеров;
- Собственно секвенирование. К кластерам добавляют праймеры, комплементарные одной из адаптерных последовательностей, с которой они связываются. Затем добавляют ДНК-полимеразу и специально модифицированные нуклеотиды с прикрепленным к ним флуорофором (разным у разных типов нуклеотидов), синтез после которых заблокирован. Они присоединяются к молекулам ДНК в соответствии с правилом комплементарности. Затем камера сканирует, какой флуорофор появился в каждом кластере по цвету свечения в лазере, и компьютер запоминает расположение кластеров. По этому определяется, какой нуклеотид встроился в цепь. После этого отщепляются флуорофоры от всех нуклеотидов, и дальнейший синтез цепи разблокируется.
Все повторяется снова: добавляются нуклеотиды, чип сканируется, чтобы определить, какой нуклеотид присоединился к какому кластеру и т.п. (рис. 13). Таким образом секвенируют до 100 нуклеотидов в каждом кластере, а всего их сотни миллионов — в итоге это десятки миллиардов пар нуклеотидов.
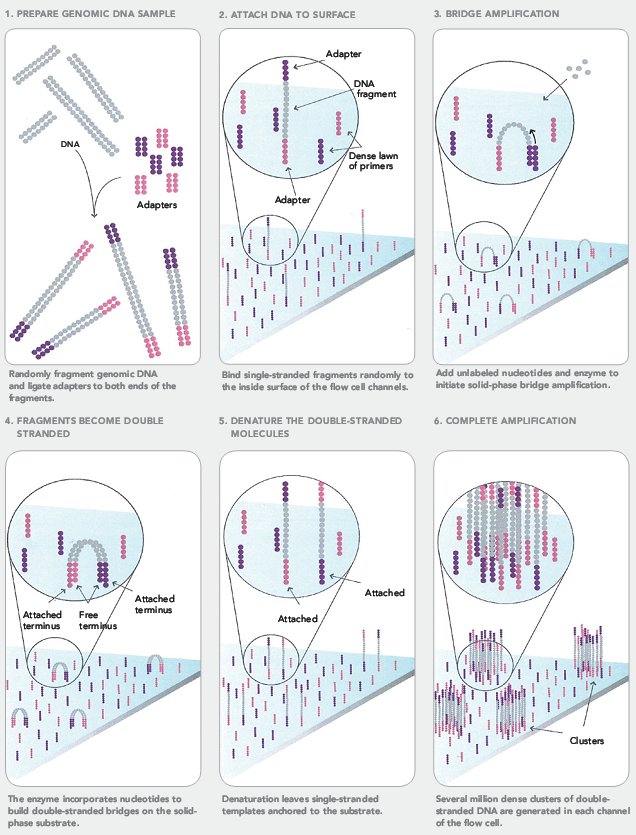
Рисунок 12. Подготовка к секвенированию Illumina.
сайт SEQanswers
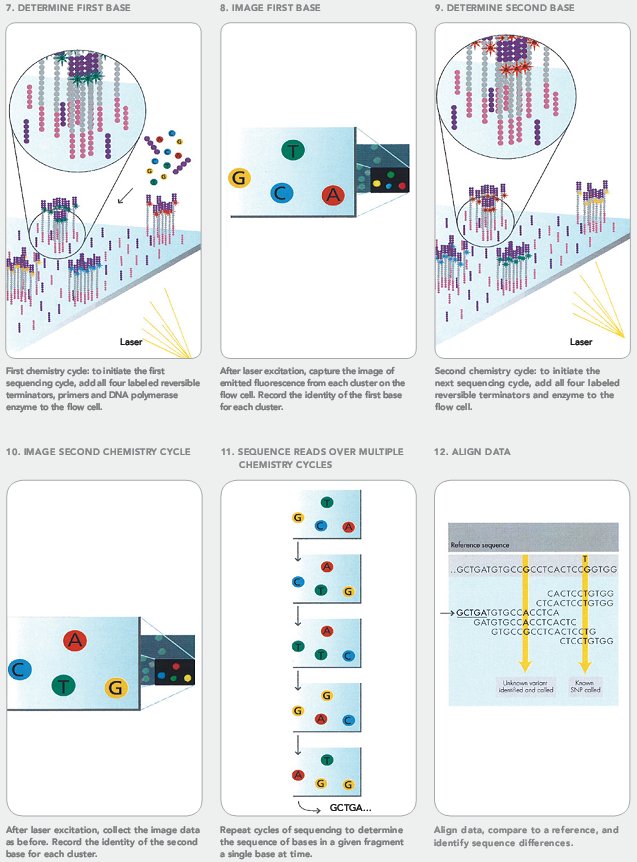
Рисунок 13. Собственно секвенирование Illumina.
сайт SEQanswers
Было уже довольно много сказано про методы работы с нуклеиновыми кислотами и их изучения. Пришло время узнать, каким образом можно выяснить, как же клетка работает — в частности, попытаться определить функцию гена и белка, который он кодирует.
In vitro-мутагенез
Для изучения функции белка очень важно научиться вносить в него мутации. Например, имея организм с неработающим ферментом, можно по биохимическим отличиям понять, что делает нормальный белок. Существуют разные способы создать полностью неработающий ген (как произвольный из всего генома, так и совершенно конкретный — тогда это называется нокаутом этого гена). Один из таких способов — вставка какого-то фрагмента ДНК в геном: если эта вставка придется на ген, то он (точнее, скорее всего, белок, который он кодирует) перестанет нормально функционировать.
Однако существуют способы очень точного изменения последовательности гена и, соответственно, белка. Про один из таких методов — сайт-специфичный мутагенез — я и расскажу. Суть его заключается в изменении конкретного (обычно одного) нуклеотида в последовательности. Для его использования сначала необходимо клонировать этот ген в плазмиде. После этого нужно провести как бы ПЦР с одним праймером. Причем этот праймер должен как раз включать в себя последовательность, которую мы хотим изменить — уже в нужном нам виде. Например, на рис. 14 вместо буквы А, которая должна была бы стоять напротив Т в родительской цепи, в праймере стоит Ц. После синтеза второй цепи ДНК плазмиды, содержащей праймер, в нее будет внесена мутация — А заменится на Ц. Такие плазмиды вводятся в клетки, в которых при делении две цепи окажутся в разных дочерних клетках. Таким образом, в половине клеток-потомков будет изначальный вариант плазмиды, а в половине — мутантный. Тогда, соответственно, половина клеток будет производить нормальный белок, кодируемый этим геном, а половина — мутантный. В случае, изображенном на рис. 14, в нем вместо одной аминокислоты (аспарагина) будет стоять другая (аланин). По аналогии можно вносить случайные мутации с помощью специальной ДНК-полимеразы, вносящей повышенное число ошибок.

Рисунок 14. Схема проведения сайт-специфичного мутагенеза.
Bruce Alberts et al. Molecular biology of the cell. 5th edition.
Системная РНК-интерференция
РНК-интерференция — недавно (менее 20 лет назад) открытый феномен подавления экспрессии генов в присутствии определенных коротких фрагментов РНК. За открытие и изучение этого явления Эндрю Файер (Andrew Fire) и Крейг Мелло (Craig Mello) получили Нобелевскую премию по физиологии и медицине в 2006 году. Биомолекула уже достаточно писала про РНК-интерференцию: «Обо всех РНК на свете, больших и малых» [15], я же расскажу о так называемой системной РНК-интерференции у «модельной» нематоды C. elegans, — то есть, об отключении гена во всех (почти) клетках этого червя.
Такой поразительный эффект достигается с помощью введения в клетку двуцепочечных молекул РНК (дцРНК), одна из цепей в каждой из которых комплементарна участку мРНК «выключаемого» гена. Это открывает поразительные возможности для изучения функций генов. Раньше для отключения генов приходилось создавать «нокаутных» животных (что ученые все равно вынуждены делать, например, с мышами — см. «Нобелевскую премию по физиологии и медицине вручили за технологию нокаутирования мышей». — Ред.), у которых изучаемый ген в принципе отсутствует в геноме. Однако создание нокаутов достаточно сложно, а обратно включить ген у таких организмов уже невозможно. С помощью РНК-интерференции отключить ген очень легко, — так же, как и включить, перестав водить в организм соответствующие дцРНК [16].
Существует три основных способа введения дцРНК в организм. Самый очевидный — впрыскивание в животное их раствора. Пользуются также «вымачиванием» нематод в растворе РНК. Однако оказалось, что можно делать все гораздо проще: скармливать нематодам эти молекулы! Причем особенно удобно то, что это так же замечательно работает, если нематод кормить бактериями (E. coli), синтезирующими эти дцРНК (рис. 15) [17].

Рисунок 15. Системная РНК-интерференция. Червь C. elegans экспрессирует зеленый флуоресцентный (светящийся) белок в клетках глотки (ph) и мышцах стенки тела (bm). Слева — изначальный внешний вид. Справа — при РНК-интерференции с помощью «подкормки» бактериями ген инактивируется.
В принципе то, что молекулы РНК из кишечника распространяются практически по всем тканям, довольно удивительно. Известно, что за попадание молекул РНК в клетки кишечника отвечает белковый канал sid-1 [18], [19]. Однако каким образом РНК распространяются по организму червя, достоверно не известно, — скорее всего, с участием белка rsd-8 [16] Интересно, что все известные белки, принимающие участие в системной РНК-интерференции у C. elegans, имеются и у человека, однако такую эффективную систему искусственного подавления активности генов на системном уровне у человека наблюдать не удается. Если бы была возможность использовать системную РНК-интерференцию у человека, это могло бы стать методом борьбы с огромным набором заболеваний, от простуды до рака .
К слову, использование РНК-интерференции именно на культуре клеток человека позволило выявить, что многие гены человека способствуют развитию вируса гриппа: «Молекулярное двурушничество: гены человека работают на вирус гриппа». — Ред.
Изучение экспрессии генов: ДНК-микрочипы
При изучении функции гена очень важно узнать, когда и в каких тканях организма он работает (экспрессируется), а также вместе с какими другими генами. Если требуется узнать это про небольшое число генов и тканей, то можно это сделать очень просто: выделить РНК из ткани, провести обратную транскрипцию (то есть, синтезировать кДНК — комплементарную ДНК) и затем, провести количественную ПЦР. В зависимости от того, прошла ли ПЦР, мы узнаем, имеется ли мРНК исследуемого гена в ткани.
Однако если необходимо проделать то же самое для множества тканей и многих генов, то эта методика становится очень долгой и затратной. В таком случае используют ДНК-микрочипы [3]. Это небольшие пластинки, на которые нанесены и прикреплены молекулы ДНК, комплементарные РНК изучаемых генов, причем заранее известно, где на них (пластинках) какая молекула расположена. Одним из способов создания чипа является синтез молекул ДНК прямо на нем с помощью робота.
Чтобы изучать экспрессию генов с помощью чипов, необходимо также синтезировать их кДНК и пометить ее флуоресцентным красителем (не разделяя кДНК разных генов). Такую смесь наносят на микрочип, добиваясь, чтобы кДНК гибридизовалась с молекулами ДНК на чипе. После этого смотрят, где наблюдается флуоресценция и сравнивают это с расположением молекул ДНК на чипе. Если место флуоресценции совпадает с положением молекулы ДНК, то в данной ткани этот ген экспрессирован. Кроме того, пометив кДНК из разных тканей разными красителями, можно изучать экспрессию сразу нескольких (обычно все-таки не больше 2) тканей на одном чипе: по цвету флуоресценции можно определить, в какой из тканей он экспрессирован (если сразу в нескольких — получится смешанный цвет) (рис. 16).

Рисунок 16. Флуоресценция на ДНК-микрочипе после обработки раствором кДНК. Всего тут примерно 37500 прикрепленных молекул ДНК.
Однако в последнее время все чаще вместо чипов используют массовое секвенирование всей кДНК из ткани (создание так называемых транскриптомов), что сильно упростилось из-за развития методов секвенирования. Это оказывается дешевле и эффективнее, поскольку знание полных последовательностей всех мРНК дает больше информации, чем просто сам факт их наличия или отсутствия.
Мы рассмотрели основные методы молекулярной биологии. Надеюсь, что вам стало немного понятнее, каким образом делаются молекулярно-биологические исследования, за что дают Нобелевские премии, и как они могут помочь в некоторых прикладных задачах. Но, более всего, я надеюсь, что вы тоже увидели красоту идей, лежащих в их основе, и, возможно, вам захотелось узнать о каких-то из этих методик подробнее.
Литература
- Нобелевские лауреаты: Дж. Уотсон, Ф. Крик, М. Уилкинс;
- Уотсон Дж.Д. Двойная спираль. Воспоминания об открытии структуры ДНК. М.: Мир, 1969;
- Bruce Alberts et al. Essential cell biology (3rd ed.). 2009;
- Википедия: «Центральная догма молекулярной биологии»;
- Roberts RJ., Vincze T., Posfai J., Macelis D. (2007). REBASE — enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 38, D234–D236;
- Википедия: Gel electrophoresis of nucleic acids (англ.);
- Википедия: Southern blot (англ.);
- Жимулев И.Ф. Лекции для студентов 3-го курса по общей и молекулярной генетике (гл. 7);
- Ребриков Д.В. ПЦР «в реальном времени». М.: БИНОМ. Лаборатория знаний, 2009;
- Телков М.В. (2006). Кари Маллис, изобретатель ПЦР. Химия и Жизнь. 8;
- R. Saiki, D. Gelfand, S Stoffel, S. Scharf, R Higuchi, et. al.. (1988). Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 239, 487-491;
- C. Ledergerber, C. Dessimoz. (2011). Base-calling for next-generation sequencing platforms. Briefings in Bioinformatics. 12, 489-497;
- 454-секвенирование (высокопроизводительное пиросеквенирование ДНК);
- Wellcome Trust: «DNA Sequencing — The Illumina Method»;
- Обо всех РНК на свете, больших и малых;
- Grishok A. (2005). RNAi mechanisms in Caenorhabditis elegans. FEBS Lett. 579, 5932–5939;
- Angelo Fortunato, Andrew G. Fraser. (2005). Uncover Genetic Interactions in Caenorhabditis elegans by RNA Interference. Biosci Rep. 25, 299-307;
- Winston W.M., Molodowitch C., Hunter C.P. (2002). Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 295, 2456–2459;
- C.P. HUNTER, W.M. WINSTON, C. MOLODOWITCH, E.H. FEINBERG, J. SHIH, et. al.. (2006). Systemic RNAi in Caenorhabditis elegans. Cold Spring Harbor Symposia on Quantitative Biology. 71, 95-100.

Комментарии


