Вылечить миодистрофию Дюшенна: конкуренция групп, единство методик
16 февраля 2016
Вылечить миодистрофию Дюшенна: конкуренция групп, единство методик
- 6363
- 0
- 8
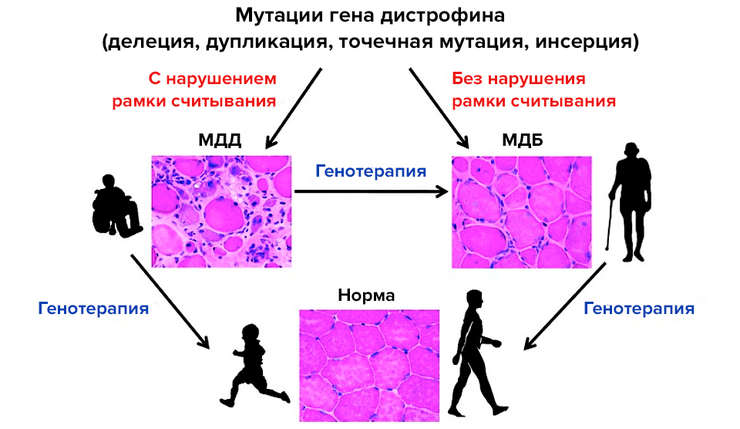
Принцип генной терапии миодистрофии Дюшенна/Беккера. Миодистрофию Дюшенна (МДД) вызывают мутации гена дистрофина (DMD), приводящие к сдвигу рамки считывания, а более мягкую миодистрофию Беккера (МДБ) — мутации без смещения рамки считывания. Лечения этой болезни пока нет. Генная терапия поможет улучшить или даже восстановить функции мышц.
[17], рисунок адаптирован
-
Автор
-
Редакторы
Мышечная дистрофия Дюшенна — тяжелейшее Х-связанное заболевание, эффективного лечения которого до сих пор нет. В одном из последних номеров Science вышли целых три статьи об успешном тестировании на мышиных моделях технологии CRISPR/Cas9 для лечения этой болезни. Может быть, у этого подхода есть шанс добраться и до клиник?
Мышечная дистрофия Дюшенна, от которой страдает один из 3600-5000 новорожденных мальчиков, вызывается отсутствием дистрофина — белка, который соединяет цитоскелет и внеклеточный матрикс в мышечном волокне и обеспечивает его стабильность при сокращении (рис. 1). Из-за мутаций гена DMD рамка считывания при трансляции его мРНК сдвигается, и синтез белка преждевременно прекращается. Врожденная болезнь очень быстро прогрессирует: ее диагностируют в возрасте около четырех лет, а к 10 годам ребенку обычно уже нужна инвалидная коляска. Это происходит потому, что без дистрофина волокна повреждаются, и как только регенеративная способность мышечных волокон исчерпывается, они заменяются фиброзной и жировой тканями [1]. Как показывают исследования, когнитивные функции у ребенка тоже могут быть нарушены [2]. Больше 30 лет с таким заболеванием, как правило, не живут, а смерть наступает от сердечных и респираторных осложнений. Более мягкий вид миодистрофии, связанной с геном DMD, — это мышечная дистрофия Беккера, когда мутации не приводят к смещению рамки считывания [3].
Дистрофин находится на внутриклеточной поверхности сарколеммы вдоль всей длины мышечных волокон и входит в состав дистрофин-ассоциированного гликопротеинового комплекса (ДАГК, DGC). Он связывается одним концом с F-актином цитоскелета, а другим — с β-дистрогликаном, что стабилизирует волокна во время сокращения. Ген дистрофина — один из самых длинных у человека.

Рисунок 1. Мутации в дистрофине — причина развития миодистрофии Дюшенна. а - Дистрофин связывается с актиновыми филаментами (часть цитоскелета) через домены N-ABD и ABD2) и с ДАГК через домены CR и CT. б — Кристаллическая структура N-ABD дистрофина. Зоны связывания с актином показаны желтым, четыре хорошо изученных мутации, вызывающих заболевание, — красным.
Излечивать мышечную дистрофию Дюшенна пока не умеют, а сегодняшняя терапия направлена на замедление прогрессирования болезни и лечение осложнений [4], [5]. «Золотой стандарт» — это кортикостероиды, которые были предложены в качестве лечения еще несколько десятилетий назад. Однако их применение вызывает множество побочных эффектов.
Неудивительно, что многие группы генетиков и молекулярщиков занимаются разработкой пре- и постнатального лечения миодистрофии Дюшенна. Болезнь в основном изучают на различных линиях мышей. В одном из последних номеров Science опубликовали сразу три независимых работы по методам лечения мышечной дистрофии Дюшенна [6–8]. Исследовательские группы возглавили Эрик Олсон (Eric Olson) из Техасского университета, Эми Уаджерс (Amy Wagers) из Гарвардского университета и Чарльз Герсбах (Charles Gersbach) из Университета Дьюка. Все группы для восстановления функции мышц использовали методику пропуска экзонов, при которой один или несколько экзонов удаляются из мРНК (рис. 2). В таком случае белок получается короче, но всё же может выполнять свою поддерживающую и заякоривающую функции в мышечном волокне, а «досадное обстоятельство» — лишний стоп-кодон — тоже оказывается «пропущенным».

Рисунок 2. Пропуск экзонов в гене дистрофина при миодистрофии Дюшенна. а — У пациентов с МДД в гене DMD присутствуют мутации, нарушающие рамку считывания при синтезе белка. Например, при делеции экзона 50 появляется «внерамочная» мРНК, что приводит к синтезу усеченного нефункционального или нестабильного дистрофина (слева). В одном из терапевтических подходов антисмысловой олигонуклеотид «маскирует» экзон 51, и он «пропускается» при сплайсинге, рамка считывания восстанавливается. В результате получается более короткий, но частично функциональный дистрофин (справа). В новых работах «лишние» экзоны просто вырезают из генома с помощью CRISPR/Cas9. б — Мультиэкзонный пропуск в терапии МДД. Если осуществить пропуск экзонов 45–55, мутации которых встречаются примерно у 63% пациентов, то образовавшийся короткий дистрофин приведёт к трансформации стандартного МДД-фенотипа в бессимптомный или более мягкий МДБ-фенотип.
У стратегии удаления экзонов есть даже преимущества перед воссозданием полной длины гена: ее проще разработать, чем восстановить индивидуальные делеции каждого пациента [7].
Для вырезания «лишних» нуклеотидных последовательностей исследователи воспользовались технологией редактирования генома CRISPR (clustered regularly interspaced short palindromic repeats) / Cas9 (CRISPR-associated protein 9) [9], которую, между прочим, только что разрешили применить в опытах на эмбрионах одному лондонскому институту [10].
Подробнее про эту методику, позаимствованную у бактерий, можно почитать в статьях: «CRISPR-системы: иммунизация прокариот», «Мутагенная цепная реакция: редактирование геномов на грани фантастики» и «А не замахнуться ли нам на ... изменение генома?» [11–13].
Конкурирующие лаборатории: кто первым воплотит технологию в терапию для человека?
Ученые трех лабораторий успешно применили технологию пропуска экзонов in vivo на стандартном объекте — мышах — и показали, что их метод помогает восстановить рамку считывания и частично восстановить синтез дистрофина. Поскольку даже невысокий его уровень (3–15% от нормального) приносит терапевтическую пользу, результаты работ можно назвать успешными.
Группа Эрика Олсона уже не в первый раз использует метод CRISPR/Cas9 в своих работах по мышечной дистрофии Дюшенна. В 2014 году ученые исправили мутацию в зародышевой линии мышей и предотвратили развитие болезни. Однако, поскольку пренатальное редактирование генома на человеческих эмбрионах (пока?) запрещено, исследователям пришлось придумать способ постнатального применения технологии.
В их последней работе для доставки необходимых для редактирования компонентов в ткани использовался аденоассоциированный вирус-9 (AAV9, adeno-associated virus-9) [6]. Исследователи испытали несколько способов введения AAV9 в различные дни после рождения мышат. Во всех случаях экспрессия гена дистрофина в сердечной и скелетных мышцах восстановилась, но в разной степени. Более того, продукция белка увеличивалась с 3 до 12 недель после инъекций, а через 4 недели после инъекций улучшилась функция скелетных мышц. «Сейчас задача для исследователей из центра Уэллстоун заключается в том, чтобы перенести открытия с мышиной модели на пациентов с миодистрофией», — говорит Прадип Маммен (Pradeep Mammen), содиректор центра Уэллстоун.
Группа Эми Уаджерс провела во многом похожий эксперимент [8]. После множества подготовительных этапов работы по редактированию генома и пропуску экзона на клетках и животных их опыт тоже увенчался успехом: программируемые CRISPR-комплексы в составе аденоассоциированного вируса (AAV) были доставлены с помощью локального и системного введения к дифференцированным скелетным волокнам, кардиомиоцитам и сателлитным мышечным клеткам новорожденных и взрослых мышей. Если редактирование направлено только на мышечные волокна, то эффект со временем может сойти на нет. Однако, как отмечает Уаджерс, редактирование генов в сателлитных клетках может обеспечить гораздо более длительный результат. Оно способно привести к созданию пула регенеративных клеток, несущих отредактированный ген дистрофина, и в результате обычной репарации мышц отредактированный ген окажется и в мышечных волокнах.
Наконец, как все уже догадались, ученые под руководством Чарльза Герсбаха тоже обнаружили терапевтический эффект применения AAV-CRISPR/Cas9 в мышиной модели [7]. Внутрибрюшинное введение вирусного вектора новорожденным мышам привело к восстановлению синтеза дистрофина в абдоминальных мышцах (мышцах живота), диафрагме и сердце через семь недель после инъекции. Как отмечают авторы, терапия сердечной и легочной мышц крайне важна, поскольку именно их отказ зачастую приводит к смерти пациентов с болезнью Дюшенна. Внутривенное введение AAV-векторов шестинедельным мышам тоже привело к значительному восстановлению продукции дистрофина в сердечной мышце. «Остается еще много работы по переделке [технологии] в терапию для человека и подтверждения ее безопасности, — говорит Герсбах. — Но результаты наших первых экспериментов уже весьма воодушевляющие». Группа собирается оптимизировать систему доставки и оценивать эффективность и безопасность стратегии на более крупных животных (рис. 3). Какая же из трех лабораторий обгонит других и первой сможет провести испытания на человеке?
Терапия миодистрофии Дюшенна: старые и новые подходы
По словам Олсона, главное отличие новой стратегии с использованием вектора, вмещающего в себя компоненты для редактирования генома, от других терапевтических методов в том, что она устраняет причину болезни. А какие еще подходы разрабатывают ученые?

Рисунок 3. Животные модели миодистрофии Дюшенна. а — Проявления миодистрофии Дюшенна у мышей и собак. Вверху: у мышей mdx симптомы проявляются только в старости, и они склонны к образованию рабдомиосарком — опухолей мышечного происхождения. Размер мышей с нокаутами генов атрофина/дистрофина и интегрина/дистрофина значительно меньше, чем их ровесников дикого типа (BL10 и BL6). Внизу: проявления болезни у пятимесячной больной собаки. Различия между здоровой и больной двухлетними собаками. б — Сравнение продолжительности жизни здоровых и больных людей, собак и различных линий мышей.
Один из многообещающих подходов — это клеточная терапия. Хотя опыты с внутримышечной инъекцией миобластов от здоровых доноров провалились, технологии с использованием стволовых клеток и индуцированных плюрипотентных стволовых клеток (ИПСК) пока успешно испытываются на моделях не только миодистрофии Дюшенна, но и болезни Альцгеймера, Паркинсона, Хантингтона, спинальной мышечной атрофии, бокового амиотрофического склероза, аутизма и шизофрении [14–16]. Например, в 2013 году исследователи из Бостонской детской больницы (Boston Children’s Hospital’s Stem Cell Program) с помощью смеси трех малых молекул (форсколина, основного фактора роста фибробластов bFGF и ингибитора гликогенсинтазы киназы-3) перепрограммировали ИПСК из кожи пациентов с миодистрофией Дюшенна в мышечные клетки, которые затем успешно прижились у мышей. Сейчас из ИПСК получены кардиомиобласты и нейроны [2].
Другие исследования показывают, что восстановление нормального уровня синтеза оксида азота (NO), который снижается у больных из-за нарушения активности NO-синтазы (nNOS), ослабляет воспаление, повышает активность собственных стволовых клеток и реконструирует морфологию и функции скелетных мышц [3].
Уже в фазе II клинических испытаний находится препарат Givinostat — ингибитор гистондеацетилаз, который замедляет прогрессирование болезни в мышиной модели.
Такой массированный экспериментальный удар по миодистрофии Дюшенна вселяет надежду. Станет ли технология CRISPR/Cas9 ведущей в разработке терапии, которую смогут принять на вооружение клиницисты? Возможно, не за горами и публикации похожих работ по другим заболеваниям, где нужно избавиться от мутаций в одном-единственном гене? Это мы узнаем из ближайших выпусков Science (а также других почетных журналов).
Литература
- van Putten M., Hulsker M., Nadarajah V.D., van Heiningen S.H., van Huizen E., van Iterson M. et al. (2012). The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One. 7, e31937;
- Russo F.B., Cugola F.R., Fernandes I.R., Pignatari G.C., Beltrão-Braga P.C. (2015). Induced pluripotent stem cells for modeling neurological disorders. World J. Transplant. 5, 209–221;
- Falzarano M.S., Scotton C., Passarelli C., Ferlini A. (2015). Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 20, 18168–18184;
- Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L. et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93;
- Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L. et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 9, 177–189;
- Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E. et al. (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 351, 400–403;
- Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M. et al. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 351, 403–407;
- Tabebordbar M., Zhu K., Cheng J.K., Chew W.L., Widrick J.J., Yan W.X. et al. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 351, 407–411;
- Элементы: «Прокариотическая система иммунитета поможет редактировать геном»;
- Gallagher J. (2016). Scientists get ’gene editing’ go-ahead. BBC News;
- CRISPR-системы: иммунизация прокариот;
- Мутагенная цепная реакция: редактирование геномов на грани фантастики;
- А не замахнуться ли нам на... изменение генома?;
- Нобелевская премия по физиологии и медицине (2012): индуцированные стволовые клетки;
- Болезнь Альцгеймера: ген, от которого я без ума;
- Как спасти Тринадцатую? (Перспективы лечения болезни Хантингтона);
- McGreevy J.W., Hakim C.H., McIntosh M.A., Duan D. (2015). Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Model. Mech. 8, 195–213;
- Singh S.M., Kongari N., Cabello-Villegas J., Mallela K.M. (2010). Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-β aggregates. Proc. Natl. Acad. Sci. USA. 107, 15069–15074;
- Goyenvalle A., Seto J.T., Davies K.E., Chamberlain J. (2011). Therapeutic approaches to muscular dystrophy. Hum. Mol. Genet. 20, R69–R78.

Комментарии


