Компьютеры против сонной болезни
08 ноября 2008
Компьютеры против сонной болезни
- 708
- 0
- 1
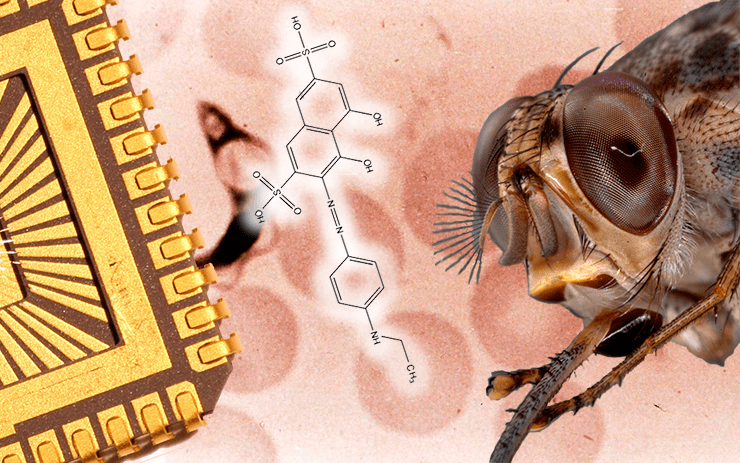
Сонную болезнь, сильнейшая эпидемия которой случилась в 1896—1906 году в Уганде и бассейне Конго, вызывает простейшее Trypanosoma brucei, переносчиком которого является африканская муха це-це. Первое лекарство от сонной болезни было предложено Паулем Эрлихом (использовалось с 1910 г.), однако, оно содержало мышьяк и было сильно токсичным.
-
Автор
-
Редакторы
Тропические заболевания, вызываемые простейшими, и, в частности, сонная болезнь (или африканский трипаносомоз), являются одной из существенных причин смертности и общего низкого качества жизни в развивающихся странах, однако эффективного лечения трипаносомоза фактически не существует. Американские учёные использовали информацию о строении жизненно важного для трипаносомы фермента митохондрий — РНК-редактирующей лигазы 1 — для компьютерной идентификации ингибиторов этого белкá, которые станут отправной точной для дизайна нового поколения лекарств против сонной болезни и ряда других заболеваний.
Беднейшие страны африканского континента подвержены заболеваниям, вызываемым простейшими и переносимым насекомыми. Среди этих болезней — африканский трипаносомоз (сонная болезнь), болезнь Чагаса и лейшманиоз. Для паразитического одноклеточного простейшего Trypanosoma brucei, вызывающего сонную болезнь, характерно чередование жизненных форм, средой обитания для которых являются по очереди кишечник насекомого (например, африканской мухи «це-це» Glossina palpalis) и кровь млекопитающих (антилоп, человека). Трипаносомоз вызывает общее истощение, анемию, повреждение разных систем органов. Через 3–6 недель после заражения наступает неврологическая стадия заболевания, связанная с разрушением клеток нервной системы и проявляющаяся в нарушении поведения и циклов гиперактивность/апатия и сон/бодрствование, за счёт чего болезнь и получила название. Эта стадия уже необратима и, как правило, заканчивается смертью. За последние 50 лет появилось только одно новое лекарство против трипаносомоза, малоэффективное и высокотоксичное.
За последнее время в биологии этих простейших было открыто много нового, в частности, отсеквенирован их геном [1] и описано уникальное для паразитических простейших, обладающих кинетопластом, явление — редактирование мРНК митохондрий [2]. Этот процесс заключается в удалении или вставке уридиновых оснований в последовательность мРНК, управляемых специальной, частично комплементарной мРНК, «руководящей» РНК, которая входит в состав сложнейших молекулярных комплексов, называемых редактосомами (по аналогии с editosome — А. Ч.). Мультиферментный ансамбль (включающий 15–20 субъединиц!) производит разрез мРНК в точке, где нарушается её комплементарность с «руководящей» РНК, и отщепляет или же, наоборот, «достраивает» основание урацила к одной из «половинок» мРНК (в зависимости от типа «рук-РНК»), после чего эти «половинки» «сшиваются» при помощи РНК-редактирующих лигаз типов 1 (REL1) и 2 (REL2). (Многие ферменты в составе редактосом — в частности, и РНК-лигазы, — являются АТФ-зависимыми.) REL1, кристаллографическая структура которой известна, жизненно необходима для всех жизненных форм трипаносомы и может поэтому быть перспективной мишенью для лекарств против трипаносомоза, тем более что близких гомологов этого белка нет в человеческом организме (а это важно, чтобы не было опасных побочных эффектов).
Группа американский учёных под предводительством известного молекулярного биофизика, одного из пионеров метода молекулярной динамики Эндрю Мак-Кэммона (Andrew McCammon) решила провести компьютерный поиск селективных ингибиторов фермента REL1, поскольку они могут стать перспективной отправной точкой для нового поколения лекарств от трипаносомоза. Используя комбинацию компьютерных алгоритмов, известных под названием молекулярная динамика (МД [3]) и виртуальное сканирование (ВС [4]) баз данных молекул, им удалось идентифицировать несколько соединений, обладающих микромолярной константой ингибирования [5].
Чаще всего для виртуального сканирования баз данных химических соединений используется метод молекулярного докинга [4], предназначенный для «вписывания» лигандов (небольших молекул) в сайты связывания на поверхности белков (ферментов или рецепторов). Можно выделить два основных результата докинга — это пространственная структура комплекса фермент-лиганд (в этом случае будет правильнее сказать «субстрат») и предсказанная энергия межмолекулярного связывания в этом комплексе, определяющая его «прочность» и, в конечном итоге, активность соединения — будь то лиганд, субстрат или, как в этом случае, ингибитор. При этом предполагается, конечно, что пространственное строение белка-мишени известно — или в результате эксперимента по определению структуры, или хотя бы в результате компьютерного моделирования [6]. В случае РНК-редактирующей лигазы 1 трипаносомы имеется экспериментально полученная структура, что и стало одним из критериев выбора этого белка в качестве «мишени».
Однако одним из наиболее принципиальных недостатков алгоритмов докинга является невозможность учёта конформационной подвижности белка-мишени. Динамическая подвижность же является неотъемлемой частью процесса связывания, и отбрасывание её может привести к абсолютно неудовлетворительным результатам. Аккуратное моделирование динамической природы белковых молекул является одной из наиболее сложных и интересных, но до сих пор в общем виде неразрешённых задач молекулярной биофизики.
Решив однако сразу, что докинг с использованием статичной структуры — это уже вчерашний день, учёные решили применить подход, называемый ими методом релаксированного комплекса, который заключается в изучении связывания лиганда не с отдельно взятой статичной конформацией, а с ансамблем конформаций, представляющим конформационное пространство белка-мишени. Набор этих конформаций был получен, конечно, с помощью широко используемого метода МД [3], с помощью которого рассчитали динамическое поведение фермента REL1 в ячейке с водой в течение 20 нс. Из этой траектории было получено 33 представительных конформации, максимально отличающихся друг от друга — это было сделано для того, чтобы, сохранив число структур минимальным (с целью экономии компьютерных ресурсов), получить представительный набор динамических конформаций REL1 в растворе. В таком подходе каждый тестируемый лиганд характеризуется не одним значением предсказанной энергии связывания, а целым спектром, медиану которого использовали для сравнения соединений между собой и ранжирования.
В качестве источника молекул для поиска активных соединений была выбрана известная база National Cancer Institute (NCI) diversity set, представляющая собой подмножество «максимально непохожих» друг на друга молекул из всего ассортимента, доступного в банке американского института NCI (не только в виде файлов, конечно, но и в виде реальных веществ). (Эта база и скомпонованные по аналогичному принципу базы часто служат начальным этапом виртуального сканирования, если даже примерно неизвестно, как должна выглядеть целевая молекула — по принципу «авось что-нибудь да найдётся».) Размер NCI diversity set сравнительно небольшой — немногим меньше 2000 молекул.
Далее с помощью «обычного» докинга в кристаллическую структуру REL1 выбрали 2% наиболее перспективных соединений (т. е., обладающих оптимальной предсказанной энергией взаимодействия с АТФ-связывающим сайтом REL1 — а именно туда должны «залипать» ингибиторы), и их уже подали «на вход» методике релаксированного комплекса. Среди восьми отобранных в результате для экспериментальной проверки эффективности ингибирования молекул, одно соединение продемонстрировало константу ингибирования около 2 μМ. Произведя выбор похожих на это молекул из «полной» базы NCI, исследователи нашли ещё 2 ингибитора с активностью того же порядка. (Поиск в «полной» базе — а это уже >260 000 соединений — осуществлялся с помощью быстрого алгоритма, вычисляющего «похожесть» по топологическим критериям, без моделирования в трёхмерном пространстве.)
Общим мотивом в строении всех трёх найденных ингибиторов является фрагмент 4,5-дигидрокси-2,7-нафталендисульфониевой кислоты (см. на рисунке в заглавии); все они относятся к тому же разряду органических красителей, что и сурамин — найдённое ещё в конце XIX века Паулем Эрлихом лекарство от паразитов, используемое и по сей день. (Правда, в состав, предложенный Эрлихом, входил ещё и мышьяк, что делало лекарство сильно токсичным.) Все открытые ингибиторы селективны к REL1, — по крайней мере, они не ингибируют ближайший человеческий гомолог ДНК-лигазу IIIβ и РНК-лигазу 2 фага Т4. (А для лекарства селективность — это очень важная характеристика.)
Это уже не первый случай, когда Мак-Кэммон и его группа применяют подобный подход для поиска веществ, на основе которых потом разрабатывают лекарства: какое-то время назад они нашли ингибиторы ВИЧ-1 интегразы, приведшие к разработке и утверждению Управлением по контролю за лекарствами и продуктами питания лекарства против СПИДа под названием ралтегравир.
Пример этой работы наглядно показывает преимущество учёта динамической природы эффекта межмолекулярного распознавания: если бы учёные руководствовались только данными «обычного докинга», описанные в работе ингибиторы скорее всего не были бы обнаружены, поскольку находились за пределами первой десятки соединений в списке докинга с использованием одной только кристаллической структуры.
Литература
- G. A. M. Cross. (2005). Trypanosomes at the Gates. Science. 309, 355-355;
- Kenneth D. Stuart, Achim Schnaufer, Nancy Lewis Ernst, Aswini K. Panigrahi. (2005). Complex management: RNA editing in trypanosomes. Trends in Biochemical Sciences. 30, 97-105;
- Молекулярная динамика биомолекул. Часть I. История полувековой давности;
- Драг-дизайн: как в современном мире создаются новые лекарства;
- R. E. Amaro, A. Schnaufer, H. Interthal, W. Hol, K. D. Stuart, J. A. McCammon. (2008). Discovery of drug-like inhibitors of an essential RNA-editing ligase in Trypanosoma brucei. Proceedings of the National Academy of Sciences. 105, 17278-17283;
- Торжество компьютерных методов: предсказание строения белков.

Комментарии


