
12 методов в картинках: очистка молекул и разделение смесей
04 августа 2017
12 методов в картинках: очистка молекул и разделение смесей
- 23176
- 2
- 43
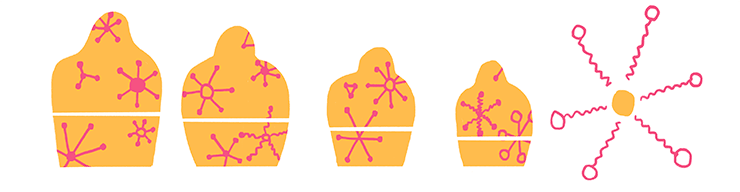
рисунок Ольги Пташник
-
Авторы
-
Редакторы
-
Иллюстратор
Все живое на нашей планете состоит из огромного количества биологических молекул — ДНК, РНК, липидов, углеводов, — объединенных в сложные системы. Для того чтобы понимать, как эти системы работают, нам надо уметь разбирать их на отдельные компоненты — так получается гораздо проще. Если вы хотите узнать, как можно подойти к анализу сложных композиций биомолекул — читайте эту статью!

12 биологических методов в картинках

Генеральный партнер цикла — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.

Партнер этой статьи — «Галахим»
«Галахим» специализируется на продаже, установке и сервисном обслуживании аналитических и препаративных хроматографов. Самый широкий в России ассортимент хроматографических колонок для ГХ и ВЭЖХ, сорбентов, пластин для ТСХ, фильтров и фильтровальной бумаги, реактивов, биохимических реагентов, микробиологических сред и стандартных образцов.
Одна из главных миссий «Биомолекулы» — докопаться до самых корней. Мы не просто рассказываем, какие новые факты обнаружили исследователи — мы говорим о том, как они их обнаружили, стараемся объяснить принципы биологических методик. Как вытащить ген из одного организма и вставить в другой? Как проследить в огромной клетке за судьбой нескольких крошечных молекул? Как возбудить одну крохотную группу нейронов в огромном мозге?
И вот мы решили рассказать о лабораторных методах более системно, собрать воедино в одной рубрике самые главные, самые современные биологические методики. Чтоб было интереснее и нагляднее, мы густо проиллюстрировали статьи и даже кое-где добавили анимации. Мы хотим, чтобы статьи новой рубрики были интересны и понятны даже случайному прохожему. И с другой стороны — чтобы они были так подробны, что даже профессионал мог бы обнаружить в них что-то новое. Мы собрали методики в 12 больших групп и собираемся сделать на их основе биометодический календарь. Ждите обновлений!
Основой жизни на нашей планете являются клетки, а каждая клетка состоит из огромного количества самых разных химических веществ — сложных и не очень [1], [2]. В химии живого есть несколько уровней сложности системы (рис. 1):
- самый нижний уровень, представленный низкомолекулярными соединениями (неорганическими и органическими ионами, аминокислотами, липидами, моносахаридами, нуклеотидами);
- средний уровень — биополимеров (белков, полисахаридов, нуклеиновых кислот) и их комплексов, включая и живую клетку как совокупность различных биологических молекул;
- высшим уровнем сложности биологической системы оказывается живой организм, составленный из множества самых различных клеток (ну или одна клетка в случае одноклеточных организмов).

Рисунок 1. «Лестница масштабов»: каждый последующий объект крупнее предыдущего в 10 и более раз.
рисунок Ольги Пташник
Для того чтобы исследовать химические процессы, связанные с жизнедеятельностью клетки и организма, необходимо уметь выделять из невероятного многообразия компонентов живого вещества то, что планируется исследовать, и уже с этим материалом работать дальше [3]. Собственно, появление вечно молодой бабушки современной молекулярной биологии — биологической химии (на заре ее становления она называлась физиологической химией), — стало возможным только после разработки методов выделения отдельных компонентов живого, для их исследования сперва in vitro, а потом и in vivo.
История создания: разделяй и властвуй
Разделение сложных смесей молекул и выделение из биологического материала отдельных его компонентов было известно человеку задолго до появления биохимии:
- разделение молока на молочную сыворотку, содержащую белок, и на сливки, содержащие преимущественно жиры-триглицериды;
- сворачивание молока сычужным ферментом в сыроварении с отделением от сыворотки практически всего белка — казеина;
- получение крахмала из зерна и клубней картофеля отмыванием его от других компонентов исходного сырья;
- выделение душистых масел из цветков, коры и плодов растений путем перегонки или экстракции жирами и маслами;
- получение жира для мыловарения вытапливанием.
Все эти практические способы были методами выделения отдельных фракций тех или иных биологических молекул из природного материала. Кратко история развития методов выделения и разделения молекул суммирована на рисунке 2.

Рисунок 2. История развития методик очистки и разделения молекул. 1833 г. — открытие амилазы (диастазы), грубая очистка из солода (А. Пайен и Ж. Персо). 1842 г. — первая книга по химии живого (Ю. Либих). 1842 г. — расщепление гемоглобина (Й. Берцелиус). 1862 г. — кристаллизация гемоглобина (Ф. Хоппе-Зейлер). 1869–1871 гг. — выделение липидов и нуклеиновых кислот (Ф. Мишер). 1877 г. — термин «биохимия» (Э. Бюхнер). 1907 г. — открытие возможности брожения сахаров в бесклеточных системах (Э. Бюхнер). 1926 г. — кристаллизация первого фермента (уреазы) (Дж. Самнер). 1926 г. — изобретение и применение ультрацентрифуги (Т. Сведберг). 1955 г. — применение электрофореза для разделения белков (О. Смитис). 1956 г. — открытие гель-хроматографии (Дж. Порат и П. Флодин) [5], [12]. Чтобы увидеть рисунок в полном размере, нажмите на него.
рисунок Ольги Пташник
Пожалуй, первое «настоящее» и хорошо документированное выделение индивидуального биологического компонента — белка гемоглобина (рис. 3) — предпринял основоположник физиологической химии, профессор Тюбингенского университета (Германия) Феликс Хоппе-Зейлер (1825–1895 гг.). В 1862 году он совершил прорывное открытие, впервые выделив из крови человека в чистом кристаллическом виде окрашенный дыхательный белок гемоглобин [4]. До этого он назывался просто «Blutfarbstoff» — «окрашивающее кровь вещество». Иенс Берцелиус (1779–1848 гг., Швеция), автор термина «органическая химия», до этого смог расщепить грубый препарат гемоглобина на органическое вещество глобулин, и на содержащий железо пигмент, способный связывать кислород.

Рисунок 3а. Гемоглобин — от структуры к кристаллу. Четыре субъединицы объединяются в компактную кубическую структуру...
рисунок Ольги Пташник

Рисунок 3б. ... — и посмотрите, как кристаллы гемоглобина снаружи напоминают молекулы, их составляющие!

Рисунок 3в. Кристаллы гемоглобина под микроскопом.
сайт images.nigms.nih.gov
Хоппе-Зейлер характеризовал полученный им белок с различных сторон — например, впервые применив ранее предложенный Бунзеном и Кирхгофом спектроскоп для описания спектров поглощения гемоглобина как в чистом виде, так и в комплексе с различными физиологически значимыми газами (кислород, углекислота, угарный газ). Далее Хоппе-Зейлер измерил способность гемоглобина к специфическому связыванию кислорода, и подтверждил современную гипотезу тканевого дыхания, согласно которой газообмен в органах осуществляется через кровь посредством гемоглобина, предложенную ранее Юлиусом Лотаром Мейером (1830–1895 гг., Германия).
Чистые препараты нуклеиновых кислот впервые выделил сотрудник Феликса Хоппе-Зейлера Фридрих Мишер (1844–1895 гг.), ранее работавший с известным немецким химиком Адольфом Штрекером (1822–1871 гг.), специалистом в области органической химии, впервые синтезировавшим аминокислоту аланин из уксусного альдегида, аммиака и циановодорода. В 1869 и 1871 годах Мишер первым указал на то, что белки и липиды являются основными химическими веществами, составляющими цитоплазму клетки, и предпринял первую попытку классификации белков — однако не преуспел в этом из-за недостаточно развитых на тот момент методов анализа. При исследовании гистологически однородной популяции лейкоцитов из гноя он впервые выделил из этих клеток вещество, названное им «нуклеин», выпадавшее в осадок при добавлении кислот и растворявшееся обратно при добавлении щелочей (рис. 4).

Рисунок 4. Микропрепарат гноя человека. Ядерный материал лейкоцитов (ДНК и белки) окрашен в розово-синий цвет красителем азур II, вокруг лейкоцитов лежат лишенные ядра эритроциты. Как можно видеть, ДНК в гное очень много. Справа — поведение «нуклеина», комплекса ДНК и белков, в растворах кислоты (выпадение в осадок) и щелочи (растворение).
рисунок Ольги Пташник
Это вещество представляло собой первый грубый препарат ДНК в истории человечества — а вернее, комплекс ДНК с белками (дезоксирибонуклеопротеид). «Согласно имеющимся гистохимическим данным, я предполагаю, что это вещество содержится в ядре клетки», — пишет Мишер, и далее фокусируется на ядре клетки — органелле, о которой тогда было известно очень мало. Нуклеин Мишера радикально отличался от всех известных на тот момент белков тем, что после осаждения не растворялся в воде, уксусной кислоте, разбавленной соляной кислоте и в растворах солей. Мишер разработал протокол выделения клеточных ядер выдерживанием клеток на холоде в растворах соляной кислоты и протокол экстракции нуклеина из ядер щелочами с последующим осаждением. После получения чистого нуклеина Мишер сделал ряд основополагающих открытий о ДНК — например, определил отношение фосфора к другим компонентам, оказавшееся весьма близким к нынешним данным (22,5% против 22,9%), установил природу ДНК как многоосновной кислоты, предположил, что нуклеин является носителем наследственных свойств [5].
Основные шаги
Со времени исторических работ первых биохимиков методы выделения и разделения различных молекул, важных для биологии, претерпели значительные изменения, но в целом их содержание осталось примерно тем же. При работе с биологическим объектом первым этапом всегда оказывается разрушение тканей и их предварительное фракционирование [3]. При этом стоит задача максимально предотвратить разрушение целевых молекул, и избавиться от максимального количества загрязняющих примесей.

Гомогенизация
Для разрушения тканей применяют различные методы — механическую гомогенизацию (например, проворачивание материала в мясорубке), гомогенизацию ультразвуком (при которой клетки разрушаются кавитацией в растворе), продавливание через небольшое отверстие под высоким давлением, замораживание-оттаивание и др. (рис. 5). В среду для разрушения тканей добавляют различные защитные реагенты — например, бета-меркаптоэтанол для предотвращения окисления белков, ингибиторы ферментов (преимущественно гидролаз, разрушающих биополимеры или меняющих их свойства), а саму работу проводят на холоде для максимального снижения скорости действия ферментов в гомогенате.

Рисунок 5. Гомогенизация. Общая схема разрушения биологического материала для получения отдельных субклеточных фракций.
рисунок Ольги Пташник

Центрифугирование
Следующим шагом разделения обычно оказывается центрифугирование (рис. 6). Дробное центрифугирование, при котором гомогенат подвергается действию всё бóльших центрифужных полей, позволяет не только отделить неразрушенные клетки, но и выделить отдельные интересующие исследователя компоненты клеток — например, ядра, митохондрии, рибосомы, мембраны. Центрифугирование существенно обогащает препарат целевыми молекулами, сильно упрощая последующие шаги по разделению сложнейшей смеси биокомпонентов, и может быть проведено для большого количества биоматериала — это очень важно для выделения минорных компонентов из смесей [6].

Рисунок 6. Центрифугирование. Вверху — устройство центрифуги. В зависимости от скорости вращения ротора и его размера центрифуга способна генерировать значительные ускорения. Роторы могут быть угловыми и бакет-роторами (с качающимися стаканами), они отличаются направлением действия центробежной силы. Внизу — схема дифференциального центрифугирования. В зависимости от силы приложенного поля мы можем получать различные фракции клетки — от самых крупных органелл (например, ядер), садящихся при небольших значениях относительного ускорения, до самых мелких (рибосом и пр.), осаждающихся большими ускорениями.
рисунок Ольги Пташник

Рисунок 7. Ультрацентрифуга. Прибор, способный создавать в пробирках с биоматериалом огромные перегрузки — до 1 000 000 g. В таком центробежном поле можно осадить даже ионы, не говоря уже про крупные белки или органеллы клетки. Центрифужный ротор сделан из особо прочного материала (титана), камера охлаждается точным холодильником (для исключения конвекции образца), ротор вращается в глубоком вакууме (для предотвращения разогрева из-за трения о молекулы воздуха). Можно сказать, что образец летит на искусственном метеорите.
рисунок Ольги Пташник
Центрифуги очень широко применяются как в промышленности (например, в молочной для отделения сливок от обрата или для разделения смесей твердых и жидких веществ), так и в лаборатории. Основные параметры центрифуги — максимальный объем вещества, помещающийся в ее ротор, и создаваемое ей центрифужное поле. Обычно это поле выражают как относительное центробежное ускорение — в единицах, кратных гравитационному показателю Земли g (ускорению свободного падения), и данная величина оказывается пропорциональна скорости вращения ротора центрифуги. Для разных целей применяют центрифуги с разными максимальными ускорениями — от сотен и тысяч g для разделения грубых смесей до сотен тысяч g для осаждения индивидуальных белков из раствора. Ультрацентрифуга (рис. 7), создающая высокие ускорения, была долгое время единственным способом определения молекулярной массы крупных белков — эта масса была пропорциональна скорости движения молекул белка в центрифужном поле.

Экстракция
После центрифугирования препарат можно подвергнуть или экстракции отдельных интересующих исследователя химически близких групп компонентов (например, смесью эфира и метанола можно количественно извлечь из мембран липиды), или предпринять дальнейшие усилия по отделению примесей. Наиболее сложным вопросом при таком разделении оказывается разделение белковых смесей — необходимо выделить зачастую весьма немногочисленный компонент смеси, при этом он не должен потерять своей естественной биологической активности. Выделенный фермент должен работать не хуже, чем он это делает в клетке, выделенный фактор транскрипции должен хорошо и специфично связываться с ДНК, иными словами, выделенные белки должны оставаться нативными — такими же, как в живой клетке.

Рисунок 8. Высаливание белка. При определенном значении рН среды белки имеют различную растворимость в растворах неорганических солей — и, повышая содержание соли в растворе, мы можем переводить определенные белки из растворенного состояния в осадок, который потом отделять центрифугированием.
рисунок Ольги Пташник
Для первичного разделения смесей белков можно применять различия растворимости белков в растворах солей. Феномен высаливания белка (рис. 8) известен давно — так, кристаллы яичного белка овальбумина добавлением насыщенного раствора сульфата аммония получил Франц Гофмейстер в 1890 году. То, что белки по-разному растворимы в растворах солей, активно используется и в промышленности — например, рассольные сыры делаются более плотными за счет засаливания в концентрированном рассоле. Если мы добавим к раствору белка насыщенный раствор хорошо растворимой соли — сульфата аммония, — достигнув, например, 10% насыщенной концентрации, то некоторые белки выпадут в осадок, то есть обратимо денатурируют. Если мы разведем этот осадок водой или не содержащей сульфата аммония буферной средой, осадок растворится, а белки из него вернутся от денатурированной к нативной конформации. Если мы будем повышать концентрацию сульфата аммония ступенчато, и всякий раз отделять осадок белка центрифугированием, мы сможем грубо фракционировать исходную смесь, а целевой белок обнаружить в определенной фракции по его свойствам, например, цвету или ферментативной активности. Именно возможность фракционирования белковых смесей высаливанием позволила разделить белки плазмы крови на фракции глобулинов (выпадающих в осадок при более низкой концентрации сульфата аммония) и альбуминов (остающихся в растворе белков меньшего, чем глобулины, размера), и именно высаливанием до сих пор пользуются для приготовления как отдельных фракций сывороточных белков для медицины, так и для выделения фракции иммуноглобулинов — антител [7].

Разделение по заряду

Рисунок 9. Изоэлектрическая точка. Заряд белковой молекулы зависит от рН среды вокруг него — когда количество отрицательных и положительных зарядов у белковой молекулы равно, ее общий заряд равен нулю. Белковые молекулы в растворе перестают отталкиваться друг от друга, начинают слипаться и могут образовать осадок. Растворимость белка в изоэлектрической точке минимальна.
рисунок Ольги Пташник
Белки и их смеси — особенно сложные для работы объектами, так как физико-химические свойства разных белков сильно отличаются друг от друга благодаря различному аминокислотному составу. Белки состоят из аминокислот, в боковой цепи которых могут встречаться полярные группы — например, кислотные или основные остатки. В зависимости от водородного показателя (рН) среды, в которой находится белок, то есть от кислотности среды, белок может иметь различный заряд, то есть белковая молекула может быть отрицательно заряжена в щелочной среде, иметь нулевой заряд в нейтральной среде и перезаряжаться (становиться положительно заряженной). Точка рН, соответствующая нулевому заряду белка, называется его изоэлектрической точкой (рис. 9), и белки в ней обычно имеют минимальную растворимость. Навязывая белку определенную кислотность среды, мы можем получить выпадение белка в осадок — и именно это заставляет скисшее молоко, в котором лактобактерии сбродили молочный сахар до молочной кислоты, разделяться на сыворотку и белковые сгустки. Изоэлектрическая точка основного белка молока, казеина, лежит как раз в области кислых значений рН.

Диализная очистка

Рисунок 10. Диализная очистка. Раствор высокомолекулярного вещества (белка, синие точки) и соли (красные точки) помещен в мешочек из целлофана (целлюлозной мембраны, поры которой пропускают ионы соли и задерживают крупные молекулы белка). Если такой мешочек поместить в раствор, не содержащий соли, то соль начнет выходить из мешочка через поры, а белок останется внутри. Примерно так устроены и наши почки — соль и низкомолекулярные продукты обмена уходят в мочу, а белки остаются в плазме крови.
рисунок Ольги Пташник
При выделении отдельных макромолекул — ДНК, белков, полисахаридов — часто встает задача отделения от них низкомолекулярных примесей. Так, например, после высаливания белка необходимо удалить из осадка избыток сульфата аммония, одновременно переведя белок в нативное растворимое состояние. Если мы поместим наш осадок в мешочек из полупроницаемой мембраны (например, целлофана — химически обработанной целлюлозы, поры которой пропускают ионы соли и другие низкомолекулярные вещества, но слишком узки для больших молекул белка или ДНК), а сам мешочек опустим в дистиллированную воду или буферный раствор с небольшим содержанием соли, осадок начнет растворяться, а соль выходить в раствор снаружи мешочка. Так будет происходить до момента, пока концентрации соли по разные стороны мембраны не сравняются. Если мы будем менять раствор снаружи, мы сможем полностью удалить все низкомолекулярные примеси из препарата — они выйдут через поры мешочка, а биополимеры останутся внутри. Такой прием называется диализом (рис. 10) — и именно по этому принципу у нас работают почки, удаляющие из организма лишние соли, продукты азотистого обмена и другие низкомолекулярные вещества, но сохраняющие в организме все высокомолекулярные белки.

Разделение по денатурации
Смесь белков иногда можно весьма эффективно разделить и способами, при которых примесные белки денатурируют (рис. 11) — превращаются в нерастворимый осадок, а целевой белок остается в растворе. Например, если мы синтезируем в клетках кишечной палочки термостабильный белок, ген которого взят из живущих в горячих источниках архей (например, фермент ДНК-полимеразу из Thermus aquaticus, температурный оптимум которого находится около 72 °С), мы можем отделить практически 99% всех белков кишечной палочки от нужного нам белка, просто прогрев гомогенат клеток при 80 градусах. Не обладающие термостабильностью белки кишечной палочки денатурируют, а нужный нам рекомбинантный белок останется в растворе.

Рисунок 11. Различные способы денатурации белков. Тепловая денатурация белков при приготовлении пищи — так еда становится вкуснее, а денатурированный белок доступнее для пищеварительных ферментов. Механическая денатурация — взбивание белков — сродни тепловой, только происходит из-за механического «запутывания» белковых молекул. Высаливание — обратимая денатурация, рассмотренная ранее.
рисунок Ольги Пташник

Гель-хроматография

Рисунок 12. Гель-хроматография. Смесь белка (синие точки) и соли (красные точки) нанесена на колонку с носителем, поры которого не пропускают белок внутрь. При промывании колонки буферным раствором соль «застревает» в носителе, свободно диффундируя в его гранулы, а белок выходит, не задерживаясь. Так можно быстро и просто разделить как смеси белков и соли, так и смеси белков разного размера (подбирая диаметр пор носителя).
рисунок Ольги Пташник
Важным способом разделения смесей биомолекул по размеру является гель-хроматография [8], или проникающая хроматография (рис. 12).
В 1959 году шведские ученые Порат и Флодин впервые предложили новый метод разделения сложных смесей белков по молекулярной массе, и это стало возможно благодаря полученному им новому материалу под торговой маркой Sephadex. Длинные линейные молекулярные цепочки полисахарида декстрана сшивали между собой так, что удавалось получить молекулярные сита — материал с контролируемым в широком диапазоне размером пор. Этот материал был доступен в виде гранул разного размера — от микрон до сотен микрон. Белки и другие биомолекулы не адсорбировались этим материалом и не теряли своей нативной активности. Материал замачивали в буферном растворе — гранулы набухали, поры заполнялись раствором и начинали напоминать маленькие поролоновые шарики.
Дальше ими наполняли длинную трубку с воронкой сверху и краном снизу, закрывали кран, и наносили сверху смесь биологических макромолекул. Крупные молекулы белка были слишком велики для того, чтобы проникнуть в поры молекулярных сит, ну а низкомолекулярные вещества (соли, витамины, коферменты, нуклеотиды) свободно диффундировали вглубь шариков сита через поры. Через воронку начинали пропускать буферный раствор, а из крана вытекал раствор, который до того был внутри шариков сита и между ними. Крупные молекулы, не вошедшие в шарики, двигались с раствором между шариками, в исключенном объеме, равном примерно объему жидкости между шариками, а низкомолекулярные вещества задерживались материалом колонки и выходили сильно позже — с объемом, примерно равным объему колонки, то есть с полным объемом. Так удалось отделить крупные молекулы от мелких (чем-то похоже на диализ, правда?), а при подборе размера пор у молекулярного сита удалось достичь и хорошего разделения смесей биомолекул на размерные классы — длинные участки ДНК от коротких, крупные белки от мелких. Этот метод до сих пор активно применяется и при определении молекулярной массы белков и ДНК — для этого колонку с носителем сперва калибруют по смеси белков или ДНК известного размера.

Хроматография

Рисунок 13. Ионобменная хроматография. Белки с различным зарядом наносят на колонку, содержащую заряженный определенным образом сорбент. Белки с противоположным заряду сорбента зарядом задерживаются в колонке, с одноименным зарядом — выходят из нее. Задержавшиеся белки можно снять с сорбента определенной концентрацией низкомолекулярной соли, растворенной в буферном растворе. Маленькие ионы соли, находящиеся в большом молярном избытке по сравнению с крупными белковыми молекулами белка, «сталкивают» их с сорбента в раствор, занимая их место.
рисунок Ольги Пташник
Если взять похожий носитель с крупными порами, но сделать его поверхность заряженной положительно или отрицательно (например, химически присоединив к носителю различные заряженные группы — аминогруппу, сульфогруппу и пр.), можно использовать его для другого вида разделения — ионообменной хроматографии [9] (рис. 13). Например, ДНК, представляющая собой поликислоту, заряженную в нейтральных или щелочных условиях отрицательно, будет взаимодействовать с положительно заряженным носителем — анионообменником, связываясь с группами на его поверхности. При добавлении в систему избытка соли ее анионы будут конкурировать с ДНК за группы на носителе, и вытеснять ДНК из комплекса с носителем, причем соли потребуется тем больше, чем длиннее молекулы ДНК, связанные с носителем. Та же картина будет наблюдаться и с белками, заряженными отрицательно. Для связывания положительно заряженных белков необходим носитель с отрицательными группами на поверхности — катионообменник. Этот метод позволяет отсеять из сложной смеси молекулы, не имеющие в данных условиях заряда, или заряженные так же, как носитель — связывание окажется невозможным, и эти молекулы просто выйдут из колонки с носителем.

Электрофорез

Рисунок 16. Гель-электрофорез. Чем молекулы ДНК объемистее, тем сложнее им бегать — все как у людей!
рисунок Ольги Пташник
Разделение смесей биомолекул в электрическом поле называется электрофорезом (рис. 16, 17, 18). Молекулы будут двигаться в сторону соответствующего полюса в поле постоянного тока, и скорость их движения будет зависеть как от степени их заряда, так и от их размера. Если мы поместим смесь биомолекул в буферный раствор и приложим постоянное напряжение, молекулы сперва начнут делиться, а потом немедленно перемешаются благодаря конвекции — водный раствор будет выполнять функции резистора, полностью рассеивая всю приложенную энергию электрического тока в виде тепла. Для предотвращения конвекции и в качестве молекулярного сита обычно используют гели агарозы (полисахарида, линейного компонента агара) или полиакриламида (полимеризованного амида простейшей непредельной карбоновой кислоты — акриловой).

Рисунок 17. Гель-электрофорез. На этом рисунке виден гель — молекулярное сито, «полоса препятствий» для заряженных молекул. Все молекулы ДНК будут двигаться по направлению к аноду, но скорость их движения будет различаться — чем молекула крупнее, тем труднее ей будет преодолевать препятствия.
рисунок Ольги Пташник
Подбирая концентрацию этих веществ, можно получать гели с заданной величиной пор, оптимизируя их для разделения тех или иных смесей. В геле биомолекулы движутся со скоростью, пропорциональной их длине — чем мельче, тем быстрее, — и заряду. Такой вариант электрофореза хорошо подходит для разделения химически однородных смесей, содержащих молекулы одного типа, но разной длины — например, смеси фрагментов нуклеиновых кислот. ДНК будет двигаться в поле постоянного тока от отрицательного полюса к положительному, и ее движение можно видеть в ультрафиолетовом свете — после добавления в гель особого красителя бромистого этидия, комплекс которого с ДНК флуоресцирует ярко-оранжевым светом. Отдельные компоненты смеси разделяются на полоски, содержащие популяцию молекул одной длины. Гель напоминает по текстуре мармелад или студень — и нужную полоску можно вырезать из геля, а молекулы из нее исследовать другими способами.
Для смесей белков такой вариант электрофореза — нативный — подходит далеко не всегда: при том, что белки состоят из аминокислот, их заряд может существенно отличаться при различных значениях рН среды, да и форма/размер белков могут варьировать в широких пределах. Представим, что мы поместили в поле постоянного тока смесь белков с положительным, нейтральным и отрицательным зарядами. Каждый из заряженных белков пойдет к своему противоположному по заряду полюсу, а нейтральные незаряженные белки останутся на месте. Более того, крупные белки могут вообще оказаться крупнее пор геля и не войти в них, несмотря на свой заряд. Для того чтобы все белки стали заряжены одноименно, и превратились в частицы примерно одинаковой формы, мы можем обработать белок анионным детергентом — поверхностно-активным веществом додецилсульфатом натрия. Именно он является основой практически всех стиральных порошков и моющих средств для бытовых нужд. Белки под его воздействием разворачиваются и становятся похожи на ниточки, плотно облепленные молекулами додецилсульфата натрия, и заряд у этих ниточек оказывается отрицательным за счет сульфогруппы детергента.
Такие белки не будут обладать нативной биологической активностью — денатурируют, но будут разделяться в геле по размерам — скорость движения таких частиц пропорциональна их длине, и все белки пробы будут двигаться в электрическом поле строго в одном направлении, от катода к аноду. Такой вариант — денатурирующий электрофорез — активно применяется для анализа белковых смесей любой сложности (рис. 18). И в нативном, и в денатурирующем электрофорезе есть задача увидеть бесцветные белки, разделенные на геле. Обычно для этого используют органические красители — например, кумасси бриллиантовый синий, плотно связывающийся с белками и окрашивающий их в синий цвет, но отмывающийся от не содержащего белки геля. Синие белковые полосы на бесцветном фоне отлично читаются, и по стандартам молекулярной массы на том же геле можно определить примерную молекулярную массу неизвестного белка.

Рисунок 18. Схема проведения белкового гель-электрофореза. Белки денатурируют бета-меркаптоэтанолом, при этом они утрачивают все виды структуры выше первичной, превращаясь в отдельные ниточки — полипептидные цепи. Эти цепи оказываются плотно покрыты детергентом — додецилсульфатом натрия, молекулы которого маскируют собственный заряд белка. Таким образом, все белки пробы будут в электрическом поле двигаться к аноду.
рисунок Ольги Пташник
Новое и новое из старого — комбинации методов

Используя примерно одну и ту же схему эксперимента — колонку, наполненную носителем, через которую можно пропускать раствор разделяемой смеси и другие растворы, — можно получить еще несколько вариантов хроматографического разделения сложных смесей. Например, если на поверхность носителя нанесены специфичные к белку антитела, белок будет связываться с ними и оставаться на поверхности носителя, откуда потом его можно снять. Такой метод называется иммунохроматографией, и это один из вариантов аффинной хроматографии — хроматографии по сродству (рис. 19). Любое вещество, связанное с носителем, и способное взаимодействовать с целевым белком — лиганд, — в принципе можно использовать для выделения целевого белка из смесей [11]. Гликопротеины (белки, содержащие углеводную часть) могут быть выделены на носителях, покрытых лектинами — белками, узнающими углеводы. Ферменты можно выделять по связыванию с иммобилизованными на носителе субстратами этих ферментов.

Рисунок 19. Аффинная хроматография. Колонка наполнена сорбентом, покрытым веществом, которое способно узнавать интересующую нас молекулу, или самими интересующими нас молекулами — смотря что мы хотим выудить из пробы. После пропускания через колонку смеси веществ в ней задержится только то, что способно специфически связаться с сорбентом. Потом это вещество можно снять с колонки или высокой концентрацией соли, или другим значением рН среды, или денатурантом — условиями, нарушающими специфическое сродство связанной молекулы и сорбента.
рисунок Ольги Пташник
Биомедицинские приложения
Практически любое исследование в биомедицинской области включает в себя то или иное разделение сложных смесей биологических молекул. Разберем диагностику инфекционного заболевания, вызванного патогенными микробами, по шагам (рис. 20).
- Взятие пробы у пациента. Биологический материал из области воспаления тщательно измельчают, смешивают с инактивирующим бактерии реагентом, а затем растворяют содержащиеся в нем клетки (патогена и пациента) так, чтобы ДНК из клеток перешла в раствор.
- Выделение ДНК. Для выделения ДНК к лизированному препарату биоматериала добавляют носитель, специфично связывающий ДНК. Носитель несколько раз отмывают от прочих компонентов пробы, не связавшихся с ним, и снимают ДНК с его поверхности.
- Амплификация ДНК. Фрагменты ДНК патогенного микроба специфически амплифицируют с помощью ПЦР, получая фрагмент ДНК определенной длины. В реакции параллельно участвует положительный контроль — ДНК патогена — и отрицательный контроль, не содержащий ДНК (он нужен для контроля чистоты реагентов).
- Электрофорез ДНК. Полученные продукты ПЦР разделяют гель-электрофорезом и анализируют результат. Наличие в продуктах ПЦР фрагмента ДНК, соответствующего ожидаемой длине и совпадающего по длине с фрагментом, полученным амплификацией с ДНК патогена (положительным контролем), подтверждает наличие ДНК патогена в исследуемой пробе.

Рисунок 20. Схема определения методом ПЦР наличия ДНК патогенного микроорганизма в пробе биоматериала пациента. Чтобы увидеть рисунок в полном размере, нажмите на него.
рисунок Ольги Пташник
В этом простейшем исследовании применяются два приема разделения смесей биомолекул — сперва для выделения ДНК, а потом для анализа продуктов ПЦР. Любой биомедицинский анализ включает в себя разделение смесей на этапе подготовки пробы, а далее на этапе получения данных. Возможны комбинации самых разных подходов к разделению смесей биомолекул, и каждый новый вариант таких систем немедленно включается в практику исследований.
Перспективы
Несмотря на почтенный возраст большинства методов разделения сложных смесей биологических молекул, эти методы нисколько не утрачивают своей актуальности. Развитие новых методов исследования, способных работать в микромасштабе, разработка новых приборов для анализа сделает разделения быстрее и удобнее, и позволит прийти к созданию новых диагностических и терапевтических приборов и подходов. Совершенству не может быть предела.
Календарь
На основе статей спецпроекта мы решили сделать календарь «12 методов биологии» на 2019 год. Эта статья представляет январь.

Литература
- Garrett R.H. and Grisham C.M. Biochemistry. Cengage Learning, 2008. — 1184 p.;
- Nelson D.L. and Cox M.M. Lehninger principles of biochemistry (6th Edition). W.H. Freeman, 2012. — 1340 p.;
- Dennison C. A guide to protein isolation. Springer Science & Business Media, 2013. — 249 p.;
- 12 методов в картинках: структурная биология;
- Ralf Dahm. (2005). Friedrich Miescher and the discovery of DNA. Developmental Biology. 278, 274-288;
- Loewen M. Ultra-centrifugation. Сайт University of the Basque Country;
- Paul Wingfield. (1998) Protein Precipitation Using Ammonium Sulfate;
- Gel filtration. Principles and methods. (2010). Handbook from GE Healthcare;
- Ion exchange chromatography. Principles and methods. (2016). Handbook from GE Healthcare Life Sciences;
- W. Clark Still, Michael Kahn, Abhijit Mitra. (1978). Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem.. 43, 2923-2925;
- Affinity chromatography. Principles and methods. (2007). Handbook from GE Healthcare;
- Richard Giegé. (2013). A historical perspective on protein crystallization from 1840 to the present day. FEBS J. 280, 6456-6497;
- Hoppe-Seyler F. (1866). Über die oxydation in lebendem blute. Med.-chem. Untersuch. 1, 133–140.
Комментарии


