12 методов в картинках: микроскопия
21 июля 2017
12 методов в картинках: микроскопия
- 28858
- 0
- 49

рисунок Ольги Пташник
-
Авторы
-
Редакторы
-
Иллюстратор
Темы
Один из старейших научных приборов — микроскоп — появился практически одновременно с наукой в ее современном виде. Этот канонический инструмент биолога более 400 лет был важнейшим средством для познания живого, и дал львиную долю наших знаний об устройстве жизни. Все это время эволюция микроскопа продолжалась, расширяя возможности увидеть неразличимое глазом.

12 биологических методов в картинках

Генеральный партнер цикла — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
Одна из главных миссий «Биомолекулы» — докопаться до самых корней. Мы не просто рассказываем, какие новые факты обнаружили исследователи — мы говорим о том, как они их обнаружили, стараемся объяснить принципы биологических методик. Как вытащить ген из одного организма и вставить в другой? Как проследить в огромной клетке за судьбой нескольких крошечных молекул? Как возбудить одну крохотную группу нейронов в огромном мозге?
И вот мы решили рассказать о лабораторных методах более системно, собрать воедино в одной рубрике самые главные, самые современные биологические методики. Чтоб было интереснее и нагляднее, мы густо проиллюстрировали статьи и даже кое-где добавили анимации. Мы хотим, чтобы статьи новой рубрики были интересны и понятны даже случайному прохожему. И с другой стороны — чтобы они были так подробны, что даже профессионал мог бы обнаружить в них что-то новое. Мы собрали методики в 12 больших групп и собираемся сделать на их основе биометодический календарь. Ждите обновлений!
История микроскопии

На пороге микромира
Собирающие (увеличивающие) линзы были известны с XI века, и очки распространились по Европе уже в XIV веке. Традиционно изобретение первого составного микроскопа приписывают отцу и сыну — Хансу и Захарию Янсенам в 1595 году (рис. 1). Этот первый микроскоп мог увеличивать изображение всего в 3–9 раз. Есть версия, что первый микроскоп создал Корнелиус Дреббель. Среди изобретателей первых микроскопов был и Галилей, создавший свой микроскоп в 1609 году. Так или иначе, ни один из изобретателей не оставил подробных описаний микромира. Микроскопия как наука началась с Роберта Гука, который в 1665 году издал Micrographia — книгу, в которой подробно описывались устройство микроскопа, основы оптики и первые наблюдения за биологическими объектами, иллюстрированные подробными рисунками [1]. Микроскоп Гука (рис. 2) состоял из трех линз и источника света — эта основа сохраняется и в современной микроскопии. Однако достичь больших увеличений удалось с помощью более простой конструкции — Антони ван Левенгук использовал, казалось бы, примитивный микроскоп всего с одной линзой (рис. 2). Однако благодаря высочайшему качеству этой линзы ему удалось достичь 200-кратного увеличения и описать клетки простейших и даже крупные бактерии. Использование всего одной линзы не создавало оптических аберраций, которые только множились при конструировании более сложной оптической системы.

Рисунок 1. Микроскопия: этапы большого пути. 1590 г. — Захарий и Ханс Янсены создают первый микроскоп. 1665 г. — первое издание книги Роберта Гука Micrographia: описание и иллюстрации первых микроскопических исследований. 1674 г. — Антони ван Левенгук с помощью своего микроскопа описывает инфузории, а в дальнейшем — бактерии, сперматозоиды, вакуоли внутри клетки и т.п. 1858 г. — Йозеф фон Герлах разрабатывает окрашивание кармином — одной из первых гистологических красок. 1878 г. — Эрнст Аббе выводит формулу Аббе, позволяющую вычислить максимальное разрешение, исходя из длины волны. 1911 г. — Оскар Хеймштадт изобретает первый флуоресцентный микроскоп. 1929 г. — Филипп Эллингер и Август Хирт конструируют эпифлуоресцентный микроскоп, в котором эффективно отфильтровывалось излучение от источника света. 1932 г. — Фриц Цернике изобретает фазовый контраст, позволяя рассматривать живые неокрашенные объекты с большим контрастом. 1933 г. — Эрнст Руска совместно с Максом Кноллем создает первый электронный микроскоп. В 1939 году с его помощью выпустили первый коммерческий электронный микроскоп. 1934 г. — Джон Маррак получает первый конъюгат антитела с красителем. Первое практическое использование Альбертом Кунсом, усовершенствовавшим технику конъюгацией с флуоресцентной меткой. 1942 г. — Эрнст Руска создает сканирующий электронный микроскоп. 1962 г. — первое описание GFP Осамой Симомурой. 1967 г. — первое использование конфокальной микроскопии Моймиром Петраном, Дэвидом Эггером и Робертом Галамбосом. 1969 и 1971 гг. — первое описание конфокальной лазерной микроскопии. 1981 г. — Герд Бинниг и Генрих Рорер создают первый сканирующий туннельный микроскоп. 1986 г. — Герд Бинниг, Келвин Куэйт и Кристофер Гербер изобретают атомно-силовую микроскопию. 1990 г. — Винфрид Денки и Джеймс Стиклер разрабатывают первый двухфотонный микроскоп. 1994 г. — Штефан Хелл: суперразрешающая электронная микроскопия на основе подавления спонтанного испускания (STED). 2006 г. — изобретение PALM/STROM-микроскопии. Чтобы увидеть рисунок в полном размере, нажмите на него.
рисунок Ольги Пташник

Рисунок 2а. Первые «ласточки». Микроскоп Гука (реконструкция).

Рисунок 2б. Первые «ласточки». Микроскоп Левенгука.

Улучшение оптики и преодоление аберраций
Оптические артефакты — аберрации — не позволяли рассматривать мелкие объекты, даже когда удавалось достичь больших увеличений. В XVIII веке некоторые мастера-оптики смогли устранить хроматические и сферические аберрации с помощью сложных оптических систем, состоящих из собирающей и рассеивающей линз из разного типа стекол с разными показателями преломления. Ахроматические линзы (то есть линзы, устраняющие хроматические аберрации) изобрел Честер Мур Халл в 1733 году. Первые ахроматические объективы для микроскопов создал Бенджамин Мартин (около 1775 г.) и значительно улучшил Йозеф Фраунгофер. Отец известного хирурга и отца антисептики Джозефа Листера — Джозеф Джексон Листер — обнаружил, что сочетание нескольких ахроматических линз позволяет исправить не только хроматические, но и сферические аберрации (1826 г.). Правда, несмотря на совершенство оптики, повышать увеличение до бесконечности нельзя: причиной тому дифракционный предел, о котором пойдет речь ниже.
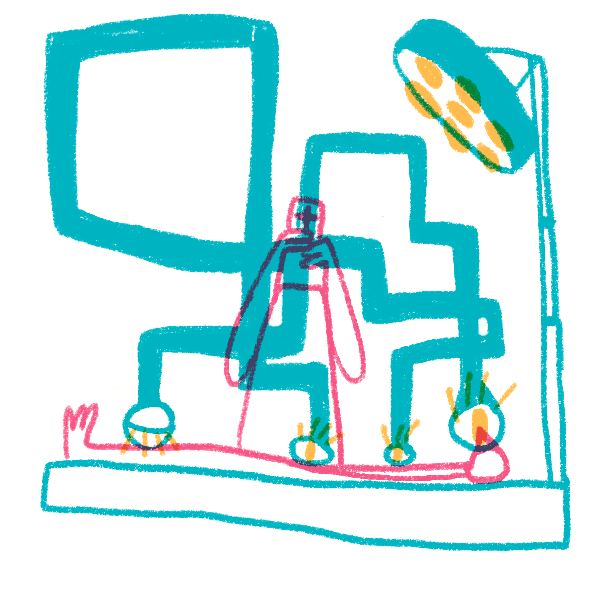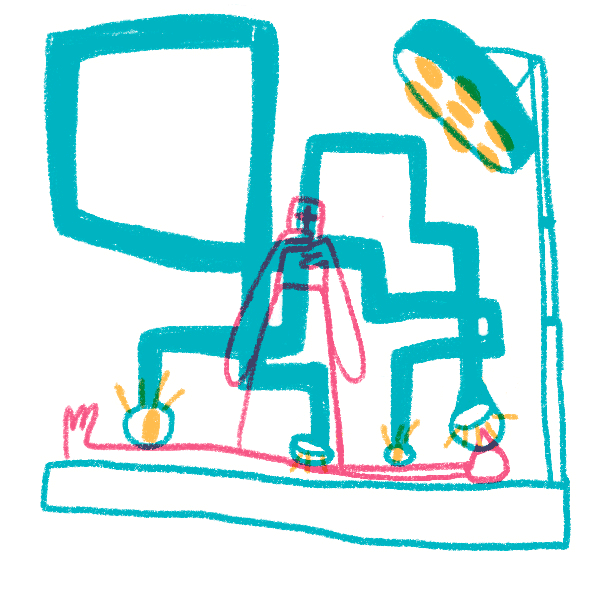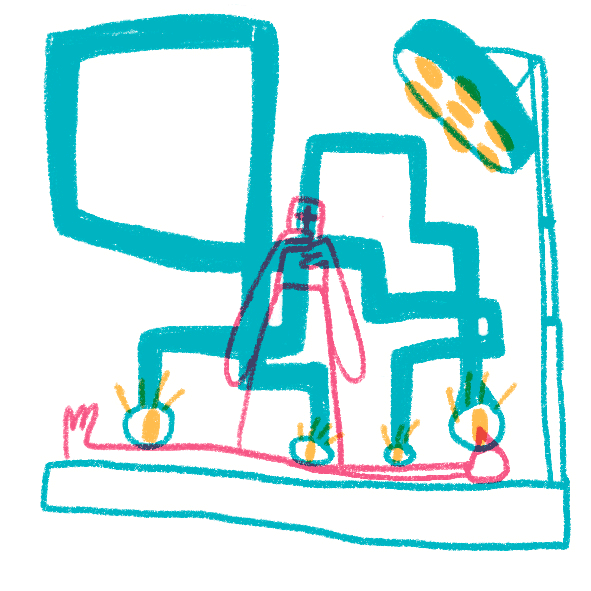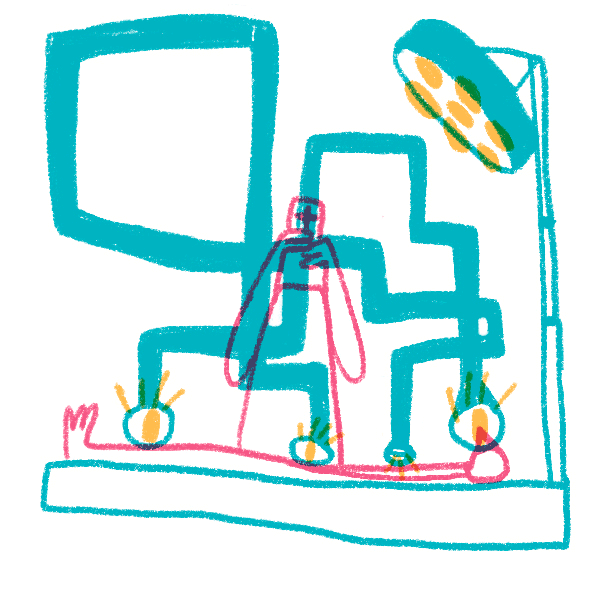
Гистология

Рисунок 3. Гистология — дело тонкое: микротом нарезает препарат на слои, прозрачные для микроскопа.
рисунок Ольги Пташник
Развитие оптики в XVIII веке позволяло исследовать микромир на больших увеличениях, однако исследование тканей и клеток было сопряжено со множеством трудностей — они же в основном не окрашены, малоконтрастны и быстро умирают вне организма. Очередной прорыв в микроскопии пришел со стороны химии. В XIX веке открыли огромное количество красителей, и биологи пробовали окрашивать ими объекты своих исследований. Одновременно изобрели микротом (рис. 3) — прибор для нарезания тонких срезов тканей определенной толщины. Для стабилизации структуры ткани выдерживали в растворах фиксаторов (например, формалина), после чего препараты хранились долгое время. Для удобства нарезания заформалиненные куски органов заключали в парафин, блоки которого удобно резать.
Ряд классических гистологических красителей, изобретенных в 19-м веке и начале 20-го, используется до сих пор: фиолетовый гематоксилин окрашивает ядра, розовый эозин — цитоплазму клеток, краситель Перлса — двухвалентное железо, реактив Шиффа — полисахариды, краситель Судан III выявляет жиры техникой Дадди, а нитрат серебра — клетки Пуркинье на срезах мозга по методу Рамон-и-Кахаля. Изобретение различных техник позволило описать тонкие структуры каждого органа, выявить входящие в них ткани и даже рассмотреть структуру клеток — ядро (с митотическими хромосомами), ядрышки и митохондрии. Микроскопические исследования того времени, хотя и были в большей степени описательными, сформировали базовые теории клеточной биологии, нейробиологии и иммунологии [2].

Математический аппарат
Дальнейшие улучшения в оптике сопровождались разработкой математического аппарата для ее описания. Джордж Биддель Эйри описал диск Эйри — дифракционный рисунок, образующийся в идеальной оптической линзе с круговой апертурой. Дифракция света ограничивает минимально возможное разрешение между двумя источниками света, и, соответственно, разрешение в оптической системе (то есть минимальное расстояние, на котором оптика микроскопа может различить раздельно две близко расположенные точки). Эрнст Аббе предложил формулу дифракционного предела в 1873 году [3]. Формула определяет предельное разрешение как:
r = λ/2NA,
где λ — длина волны источника света (от 390 до 700 нм), а NA — числовая апертура объектива, определяемая как:
NA = n×sin(θ),
где n — индекс преломления среды между объективом и препаратом, а θ — апертурный угол между крайними лучами конического светового пучка, попадающего в объектив.
Из формулы видно, что разрешение тем выше (то есть значение r меньше), чем меньше длина волны (синий свет предпочтителен красному) и чем больше числовая апертура и индекс преломления среды. Поэтому для увеличения разрешения микроскопа используют масляные объективы, когда между препаратом и объективом добавляется иммерсионное масло с высоким показателем преломления. Более строгим является критерий Рэлея, в котором предел разрешения определяется как:
R=0,61×λ/NA
Разница с пределом Аббе вызвана отличием в том, как два оптика определяли разрешение между двумя точками (критерий Рэлея строже).
Устройство микроскопа

Рисунок 4. Под увеличительным стеклом: принципиальное устройство микроскопа. Оптический микроскоп создает огромные увеличения с помощью увеличивающих линз объектива и окуляра. Промежуточные линзы устраняют аберрации, а яркий источник света позволяет рассмотреть препарат даже при больших увеличениях.
рисунок Ольги Пташник
Принципиальное устройство светового микроскопа мало менялось со времен Аббе, и основные улучшения касались уменьшения аберраций за счет усложнения состава и количества линз. Оптический микроскоп состоит из источника света, объектива и окуляров. Препарат, освещенный светом лампы, должен находиться немного дальше фокуса объектива, что создает увеличенное изображение, которое усиливается и проецируется линзой окуляра и хрусталиком глаза на сетчатку (или на матрицу камеры). В микроскопах с «бесконечной» оптикой (не ограниченной определенной длиной тубуса) препарат находится в фокусе объектива, и для построения промежуточного изображения необходима дополнительная тубулярная линза. В современных микроскопах каждый из этих компонентов включает в себя большое количество сложных линз для коррекции хроматических и сферических аберраций, а также диафрагмы, позволяющие регулировать размер поля зрения и интенсивность света. Исследовательские микроскопы оснащены несколькими объективами с разным увеличением (от 2,5—4× до 100×), которые имеют одинаковую фокусную длину и могут свободно меняться с помощью револьверной насадки. Таким образом можно найти интересный участок препарата, используя небольшое увеличение, позволяющее рассмотреть широкое поле зрение, и затем разглядеть подробности, сменив объектив на более мощный.

Рисунок 5а. Микроскопические изображения окрашенных срезов тканей. Эластичный хрящ уха (окраска: гематоксилин-эозин).
сайт www.flickr.com

Рисунок 5б. Микроскопические изображения окрашенных срезов тканей. Срез придатка яичка (окраска: гематоксилин-эозин).

Рисунок 5в. Микроскопические изображения окрашенных срезов тканей. Толстая кишка крысы (пентахромная окраска по Мовату).
сайт ru.pinterest.com
Линза окуляра дополнительно увеличивают промежуточное изображение в 10–25 раз, создавая огромные увеличения. Световая микроскопия позволяет хорошо рассмотреть тонкие препараты (например, срезы ткани или клетки, растущие на стекле), окрашенные цветными красителями (например, антителами с цветной меткой или гистологическими красителями) (рис. 5). Окраска препарата требует его химической фиксации, что не подходит для наблюдения за живыми объектами, а поэтому в большинстве современных микроскопов вводят дополнительные элементы, позволяющие наблюдать микроскопические объекты без фиксации — с помощью методов фазового контраста и его модификаций.

Фазовый контраст, видеомикроскопия и клеточные культуры
В то время как световая микроскопия подошла к своему физическому пределу, возникло два новых типа микроскопии, значительно расширяющих возможности метода. Одной из таких модификаций стал фазовый контраст.
Статичность классической гистологии не позволяла наблюдать процессы, происходящие в клетках, в реальном времени. Уже в 19 веке начались первые попытки наблюдать за тканями вне организма. Используя растворы соли и кровь или сыворотку животных, исследователи могли некоторое время поддерживать ткани в живом состоянии вне организма. В 1907 году Росс Гаррисон наблюдал развитие нервных волокон в течение недели. Использование асептических условий, протеолитических ферментов и культуральных сред позволяло выращивать отдельные клетки в течение долгого времени, проводить с ними эксперименты и, конечно, наблюдать с помощью микроскопа. В том же 1907 году Джулиус Риес снял первый микроскопический фильм — развитие эмбриона морского ежа (рис. 6) [4]. В 1913 и 1914 годах Жан Командон и Жустин Жолли снимали деление клеток в клеточной культуре. Изобретение фазового контраста Фрицем Цернике в 1941 году позволило наблюдать неокрашенные биологические объекты — прежде всего живые клетки в культуре. Фазовый контраст и его вариация — дифференциальная интерференционно-контрастная микроскопия (оптика Номарского) — стали основой как для рутинного наблюдения за клеточной культурой, так и для изучения клеточной подвижности и динамики движения органелл.

Рисунок 6. Снято! Раскадровка первого микроскопического фильма: развитие эмбриона морского ежа.

Рисунок 7. Фазовый контраст — микроскопия контрастов. Использование интерференции позволяет увеличить контрастность и изучать неокрашенные препараты. Свет, не провзаимодействовавший с препаратом, попадает на фазовую пластинку, которая смещает его на +1/4 длины волны, в то время как биологические объекты рассеивают свет так, что он не попадает на фазовую пластинку, и смещают его фазу на −1/4 длины волны. Получающаяся разница в 1/2 длины волны в тех местах, где свет проходит через препарат, приводит к тому, что в этих местах волны прямого и дифрагированного света взаимно гасят друг друга, создавая темные, контрастные области на изображении.
рисунок Ольги Пташник
Фазовый контраст основан на интерференции волн света — две световых волны, фазы колебаний которых совпадают, усиливаются, в то время как при отличии фазы на 1/2 длины волны происходит полное гашение. Проходя через материал с большим индексом отражения, свет замедляется, что приводит к смещению его фазы. Это и используется в методе фазового контраста для увеличения контрастности изображения (рис. 7). В оптический путь световой микроскопии вводятся два дополнительных элемента — обычный объектив заменяется на объектив с фазовой пластинкой, а в конденсор осветителя добавляется кольцевая диафрагма, в которой свет может пройти только через тонкое кольцо.
Кольцевая диафрагма блокирует бóльшую часть света, оставляя только полый конус света, сконцентрированный конденсором на препарате. Свет, не рассеянный и отраженный препаратом, точно попадает на фазовую пластинку объектива — напыленное полупрозрачное кольцо, которое смещает фазу света на +1/4 длины волны. Свет же, прошедший через биологические объекты, рассеивается и отражается, отклоняясь от оптического пути и не попадая на фазовое кольцо объектива. К тому же биологические объекты сами меняют фазу света за счет разницы в индексе отражения и толщины препарата, чаще всего приблизительно на −1/4 длины волны. Таким образом, между рассеянным препаратом светом и светом, не провзаимодействовавшим с препаратом, создается разница фаз в 1/2 длины волны. Рассеянный свет от препарата и свет, попавший на фазовую пластинку, снова совмещаются, приводя к ослабляющей интерференции, и, соответственно, затемнению мест препарата, отличающихся по толщине и индексу отражения.

Флуоресцентный микроскоп и иммунофлуоресценция
В 1911 году Оскар Хеймштадт разработал первый флуоресцентный микроскоп, в котором мощный источник света возбуждал свечение во флуоресцентных веществах. В 1929 году его конструкцию значительно улучшили Филипп Эллингер и Август Хирт, которые создали микроскоп, где с помощью светофильтров эффективно отсекался возбуждающий свет, что позволяло рассмотреть даже слабую флуоресценцию [5]. В это время биохимики и генетики уже начинали исследовать отдельные белки и гены, но в микроскопии не было инструмента, который бы позволил точно пометить отдельный белок. Таким инструментом удивительным образом оказались антитела — белки, которые иммунная система использует для выявления и нейтрализации возбудителей [6]. Джон Маррак одним из первых связал антибактериальное антитело с красителем (R-солью) и окрасил им бактериальный препарат. Изобретение флуоресцентного микроскопа и связывание антител с яркими флуоресцентными красителями позволило получать ярко окрашенные препараты. Меченые антитела помогали окрасить не только возбудителей болезней — иммунизируя животных с помощью различных белков, стало возможным получать антитела для выявления местоположения и количества этих белков внутри клеток и тканей (рис. 8) [7].

Рисунок 8а. Возможности флуоресцентной микроскопии. Клетки остеосаркомы человека (окраска на митохондрии, ДНК и актин).

Рисунок 8б. Возможности флуоресцентной микроскопии. Клетки бычьего эндотелия BPAEC (окраска на митохондрии, ДНК и актин).
сайт www.thorlabs.com

Рисунок 8в. Возможности флуоресцентной микроскопии. Стадии митоза (синий — ДНК, зеленый — веретено деления, красный — центромеры).
сайт wellcomeimages.org
Для того чтобы наблюдать за белками вживую, требовалось найти способ прикрепить к ним светящуюся молекулу — флуорофор. Первым таким флоурофором оказался знаменитый зеленый флуоресцентный белок (GFP). Впервые GFP выделил из медузы Aequorea victoria Осаму Симомура в 1962 году [8]. Затем зеленый флуоресцентный белок с помощью генетической инженерии научились вставлять в клетки и включать в состав белков-химер, где его «сшивали» с изучаемым белком (рис. 9). Помимо этого довольно быстро обнаружили белки всех цветов спектра [9], а их свойства усовершенствовали для лучшей флуоресценции и стабильности. За флуоресцентные и генетические технологии, связанные с GPF, в 2008 году вручена Нобелевская премия [10]. Сегодня есть флуоресцентные белки, которые светятся в присутствии определенных молекул (кальция [11], АТФ, глюкозы, глутамата и т.п.), начинают флуоресцировать при связывании двух белков друг с другом, активации определенных клеточных процессов (например, клеточной гибели) или изменении активности транскрипционных факторов.

Рисунок 9. «Да будет свет»: флуоресцентные белки помогают наблюдать процессы в живых клетках. С помощью генетической инженерии ген GFP совмещается с геном белка, за которым мы хотим «установить слежку», и получившийся химерный ген кодирует белок, помеченный с помощью GFP. С помощью этого подхода можно пометить клетки, экспрессирующие нужные нам гены, или наблюдать за биохимическими реакциями и взаимодействиями белков. Чтобы увидеть рисунок в полном размере, нажмите на него.
рисунок Ольги Пташник

Рисунок 10. Флуоресцентный микроскоп: лабиринт отражений. Дихроичное зеркало и объектив фокусируют возбуждающий свет на препарате и направляют флуоресцентный свет к окуляру по другому оптическому пути.
рисунок Ольги Пташник
Флуоресцентная микроскопия позволяет получать красочные изображения, светящиеся собственным светом, и в то же время за счет использования светофильтров рассматривать свечение разных цветов отдельно. Так можно изучать распределение нескольких меток, например антител к разным белкам, каждое из которых окрашено своим флуоресцентным красителем. В основе метода лежит Стоксов сдвиг: при возбуждении электрона во флуоресцентном красителе (флуорофоре) источником света, испускаемый фотон обладает большей длиной волны за счет потери части колебательной энергии, что позволяет отфильтровать яркий свет, которым освещают образец для возбуждения, и наблюдать только свет от флуоресцентной метки (рис. 10).
Источник света во флуоресцентном микроскопе — яркая лампа (ртутная или галогеновая). Свет от лампы проходит через светофильтр, который оставляет оптимальные для возбуждения флуоресцентной метки длины волн. Прошедший через светофильтр свет отражается дихроичным зеркалом и фокусируется объективом на образце, возбуждая флуоресценцию метки. Такая схема микроскопа, в которой возбуждающий свет фокусируется не конденсором, а самим объективом, называется эпифлуоресцентной, и позволяет значительно уменьшить фоновую засветку от лампы. Флуоресцентный свет от препарата через объектив вновь попадает на дихроичное зеркало, которое уже прозрачно для смещенной длины волны света от флуоресцентной метки, и на второй светофильтр, который дополнительно отсекает фоновый свет от лампы. Микроскоп содержит несколько комплектов светофильтров и зеркал, которые можно быстро сменить в зависимости от метки, что позволяет снимать даже микроскопические фильмы с несколькими флуоресцентными метками одновременно.
Современные микроскопы позволяют изучать препарат всеми вышеописанными методами, быстро заменяя или совмещая компоненты оптического пути, например, фазового контраста и флуоресценции. Быстрое последовательное переключение между флуоресценцией и фазовым контрастом (например, за счет автоматического открытия диафрагмы) позволяет получить обоими методами фотографии одного и того же участка препарата, которые затем можно совместить для получения суммарного изображения. Такой микроскоп дает максимум информации на одном и том же препарате за небольшое время.

Конфокальная микроскопия
Качество флуоресцентной микроскопии было ограничено из-за возбуждения флуоресценции вне оптического фокуса. Конфокальная микроскопия позволила рассматривать флуоресценцию только в фокусе объектива, и реконструировать трехмерные изображения из послойно снятых фокусных планов. Для этого используют точечный источник света, сканирующий образец, и точечную диафрагму, отсекающую свет вне оптического фокуса [12]. Первую такую конструкцию запатентовал Марвин Мински в 1961 году. Однако первый работающий микроскоп сконструировал в Чехословакии Моймир Петран (рис. 11), который в 1967 году вместе с Дэвидом Эггером и Робертом Галамбосом впервые применил его для изучения биологических образцов. В 1980-х годах в качестве источника света начали применять лазер, и конфокальная микроскопия быстро стала альтернативой обычному флуоресцентному микроскопу.

Рисунок 11. Моймир Петран. Чехословацкий ученый с коллегами создал первый работающий конфокальный микроскоп.
сайт vyznamneosobnosti.cz

Рисунок 12. Конфокальная микроскопия: через замочную скважину. В конфокальной микроскопии свечение возбуждается лазером, ограниченным точечной диафрагмой. Вторая диафрагма, помещенная после дихроичного зеркала и светофильтра эмиссии в плоскости фокуса объектива (конфокальной плоскости), отсекает свет, находящийся не в фокусе объектива. Передвижение луча лазера по препарату достигается двумя зеркалами, контролируемыми точными гальванометрическими двигателями. Переход между оптическими плоскостями (ось Z) осуществляется с помощью движения объектива вверх или вниз. Чтобы увидеть рисунок в полном размере, нажмите на него.
рисунок Ольги Пташник
Лазерная сканирующая конфокальная микроскопия является модификацией флуоресцентной микроскопии, улучшающей качество изображения и позволяющей строить трехмерные реконструкции препарата, а также в некоторых модификациях манипулировать биологическими объектами с помощью лазера. В конфокальной микроскопии, в отличие от флуоресцентной, свечение возбуждается не во всём препарате, а точечно. Перемещение лазера сканирует препарат, подобно тому, как распечатывает изображение принтер.
Соответственно, по сравнению с флуоресцентной микроскопией, в конфокальном варианте появляется несколько новых компонентов (рис. 12): прежде всего это две точечные диафрагмы, отсекающие свет, который находится вне фокуса объектива. Возбуждение света происходит с помощью лазера, сканирующего препарат, а регистрация — чувствительным фотоумножителем. Конфокальный микроскоп позволяет получать тонкие оптические срезы, и так как флуоресценция от препарата вне фокуса не попадает на детектор, то им можно снимать объемные объекты — например, стадии эмбрионального развития (рис. 13). Так как интенсивность лазера можно регулировать, а точечная диафрагма концентрирует свечение в одной точке, конфокальный микроскоп позволяет использовать лазер для точного воздействия на объект исследования. Например, лазером можно выжечь свечение флуоресцентного белка в части клетки и замерить скорость его восстановления в этой области, то есть скорость, с которой происходит диффузия белка. Этот метод называется FRAP (Fluorescence recovery after photobleaching). С помощью лазера можно повреждать клетку или только ее ядро, или активировать светочувствительные белки, запуская различные клеточные процессы.
Двухфотонная (мультифотонная) микроскопия является модификацией конфокальной микроскопии, позволяющей отказаться от точечных апертур [13]. Возбуждение в таком микроскопе происходит за счет возбуждения флуорофора одновременным попаданием двух фотонов неоптимальной длины волны от мощного лазера. Вероятность такого события падает с квадратом расстояния, поэтому эффективное возбуждение происходит только в месте фокусировки лазера.

Рисунок 13а. Конфокальная микроскопия: не только срезы, но и сравнительно крупные объекты. Периферийные нервы в зародыше мыши.

Рисунок 13б. Конфокальная микроскопия: не только срезы, но и сравнительно крупные объекты. Гиппокампальные нейроны на срезе мозга крысы.

Рисунок 13в. Конфокальная микроскопия: не только срезы, но и сравнительно крупные объекты. Окраска мышиной сетчатки на маркер астроцитов (красный — GFAP) и кровеносные сосуды (зеленый — коллаген IV).

Электронная микроскопия
Дифракционный предел больше ста лет ограничивал оптическую микроскопию, пока не появились идеи, как его преодолеть. В 1930-х годах создали новую технику, позволяющую рассмотреть внутриклеточные структуры максимально близко: электронную микроскопию. Ее идея состоит в использовании электронов в качестве средства «освещения». Длина волны у электрона на 5 порядков (в 100 000 раз) меньше, чем у фотона, что дает разрешение до 50 пикометров! Электронная микроскопия позволяет рассматривать вирусы и структуру белковых комплексов.
В принципе, устройство электронного микроскопа очень похоже на микроскоп оптический. В качестве источника электронов (осветителя в обычном микроскопе) используется электронная пушка, в которой поток электронов от источника (например, вольфрамовой нити) фокусируется с помощью цилиндрического электрода (цилиндра Венельта). В качестве линз используют электромагнитные катушки, работающие как магнитные линзы. Для того чтобы электроны не задерживались молекулами воздуха, внутри микроскопа создают вакуум.

Рисунок 14. Принцип работы трансмиссионного электронного микроскопа. Электроны от пушки фокусируются магнитными линзами и, проходя через сверхтонкий образец, попадают на флуоресцентный экран.
рисунок Ольги Пташник

Рисунок 15. Сканирующий электронный микроскоп позволяет изучать поверхность препарата за счет многих видов отраженного излучения.
рисунок Ольги Пташник
В трансмиссионной электронной микроскопии (рис. 14) изучают очень тонкие срезы клеток: препараты вирусов и тому подобные неплотные образцы. Часть электронов рассеивается или поглощается образцом, в то время как прошедшие через образец электроны попадают на флуоресцентный экран, создавая «тень» от образца — где более плотные части образца будут более темными на изображении. Трансмиссионная электронная микроскопия требует предварительной фиксации и обезвоживания препарата, а также его очень тонкой нарезки, и часто для увеличения контрастности препарат необходимо окрасить соединениями тяжелых металлов. Все эти процедуры не дают возможности наблюдать живую клетку и часто искажают изображение препарата. К тому же подготовка препаратов крайне трудоемка по сравнению с оптической микроскопией.
Электронная микроскопия позволяет метить молекулы с помощью антител, используя металлические метки (например, коллоидное золото) вместо флуорофоров, однако количество меток, которое можно использовать, ограничено по сравнению с флуоресцентной микроскопией. Несмотря на то, что именно трансмиссионная электронная микроскопия позволила нам рассмотреть структуру органелл внутри клетки, ее ограничения и дороговизна не позволяют этому методу войти в широкий лабораторный обиход.
Используя пучок электронов, образец можно «сканировать» поточечно (рис. 15), как в конфокальной микроскопии. Такой метод называется сканирующей электронной микроскопией, и в ней обычно регистрируются вторичные электроны, испускаемые образцом при попадании электронного пучка, и электроны, отраженные образцом. Метод сканирующей электронной микроскопии обычно используют для изучения поверхности образца, хотя вариант сканирующей трансмиссионной электронной микроскопии позволяет изучать тонкие срезы.
Именно сканирующая электронная микроскопия дает потрясающие объемные изображения клеток, бактерий, кристаллов, белковых матриксов и многих других объектов (рис. 16).

Рисунок 16а. Трансмисионная электронная микроскопия. Структура цитоскелета около ведущего края фибробласта.

Рисунок 16б. Сканирующая электронная микроскопия. Макрофаги и эндотелиоциты.

Рисунок 16в. Сканирующая электронная микроскопия. Фрагмент глаза дрозофилы.

Рисунок 16г. Сканирующая электронная микроскопия. Апоптотическая клетка.
сайт www.anatomybox.com

Методы суперразрешения
Электронная микроскопия продвинула исследования далеко за предел разрешения оптической микроскопии, однако она не позволяет смотреть на живые объекты и использовать разноцветные метки или красители. Возможно ли преодолеть дифракционный предел в оптической микроскопии? Методы, преодолевающие предел Аббе, называются методами суперразрешения. Мы подробно остановимся на двух таких методах, за которые присудили Нобелевскую премию по химии 2014 года [14].
В 1990-х исследователи начали разрабатывать методы для преодоления дифракционного предела Аббэ. Центральную роль в разработке этих методов играл Штефан Хелль, который разработал STED-микроскопию [14–16]. В методике STED возбуждение флуорофора происходит с помощью двух лазеров — один возбуждает флуоресценцию в точке фокуса, в то время как другой тушит ее вокруг этой точки за счет смещения флуоресценции в более красную область. За достижением Хелля последовало изобретение других суперразрешающих методов — PALM/STORM, GSDIM и других.
Штефан Хелль одним из первых понял, что возможно преодолеть дифракционный предел, используя свойства самих флуорофоров. Идея Хелля состояла в том, что не обязательно ограничивать дифракцию света, который стимулирует флуоресценцию, если можно «запретить» флуоресцировать молекулам вне самого центра фокуса. Для этого он придумал использовать принцип стимулирования испускания: в нем флуорофор в возбужденном состоянии переводится в невозбужденное состояние вторым лазером. За счет использования «более красной» длины волны испускаемый при стимулированном переходе фотон имеет большую длину волны (такую же, как и лазер) и не регистрируется детектором.
Как же можно увидеть изображение, если мы подавили флуоресценцию во всем образце? Дело в форме луча света второго лазера — проходя через специальную фазовую пластинку, он приобретает форму кольца, и интенсивности подавляющего лазера в узкой центральной точке не хватает для подавления флуоресценции. Таким образом, флуоресценция возбуждается только в узкой фокусной точке, которая по размерам гораздо меньше предела дифракции. STED-микроскопия (рис. 17 и 18) позволяет получать изображения с разрешением меньше 20 нм в поперечном направлении, и около 40–50 нм в продольном направлении.

Рисунок 17. Принцип микроскопии суперразрешения. Для STED-микроскопии необходимы два лазера: флуоресценция возбуждается первым зеленым лучом, а второй, красный, лазер подавляет флуоресценцию за счет стимуляции испускания. Из-за фазовой пластинки в середине пути второй лазер имеет форму кольца, поэтому в центре его мощности не хватает для подавления флуоресценции. Таким образом флуоресценция возбуждается только в центре — в узкой фокусной точке, куда попадает свет только от первого лазера и которая по размерам гораздо меньше предела дифракции. Чтобы увидеть рисунок в полном размере, нажмите на него.
рисунок Ольги Пташник

Рисунок 18а. Преимущества микроскопии суперразрешения. а — Ядерная пора («обычная» конфокальная микроскопия vs. STED). б — Цитоскелет («обычная» конфокальная микроскопия vs. STED).
сайты www.photonics.com и www.picoquant.com

Рисунок 18б. Преимущества микроскопии суперразрешения. STORM-микроскопия.
Другим подходом стало ограничение флуоресценции флуорофоров по времени. В PALM/STORM/FPALM-микроскопии используют специальные флуорофоры (флуоресцентные белки или синтетические флуорофоры), которые могут переключаться между возбужденным и невозбужденным состоянием (см. видео) [17], [18]. Низкочастотный возбуждающий лазер возбуждает лишь часть молекул, так чтобы статистически возбужденные молекулы были разделены расстоянием больше дифракционного предела. Высокочувствительная камера регистрирует флуоресценцию от этих молекул, позволяя точно определить их положение по испускаемым фотонам. Второй лазер подавляет флуоресценцию и возвращает молекулы обратно в невозбужденное состояние. После многократного повторения этого цикла (в каждом цикле случайно возбуждаются разные молекулы флуорофора), компьютер составляет цельную картину из изображений, снятых в отдельных циклах.
Видео. PALM-микроскопия
STED и PALM/STORM являются наиболее распространенными видами суперразрешающей микроскопии, однако существует множество методов на их основе или использующих сходные принципы: SIM, GSD, SSIM, dSTROM и другие — оптическая микроскопия продолжает развиваться, расширяя границы нашего познания о микромире.
Применение в биомедицине
Даже обычные световые микроскопы до сих пор активно используют для постановки диагнозов в медицине. Опухоли, воспалительные процессы, отложения железа или кальция, замещения нормальной ткани соединительной и многое, многое другое рутинно проверяется на препаратах, полученных с помощью биопсии, постоперационно или во время вскрытия. Широкое применение антител проникло и в рутинную диагностику — срезы окрашивают антителами к клеточным маркерам или возбудителям болезней, которые позволяют уточнить или подтвердить диагноз и лучше подобрать лечение. Особенно широкое применение световая микроскопия и иммуногистохимия нашла в онкологии, где присутствие специфических белков определяет применяемую химиотерапию (например, применение герцептина для опухолей с белком HER2 при раке молочной железы). Микроскопия необходима не только для постановки диагнозов, но и в процессе лечения: хирурги применяют микроскопы при операциях, требующих тонкого вмешательства, — в офтальмологии, нейрохирургии, сосудистой хирургии и многих других (рис. 19).

Рисунок 19а. Многообразие микроскопов в медицине. Хирургический микроскоп.
сайт redstarvietnam.com

Рисунок 19б. Многообразие микроскопов в медицине. Микроскопическая платформа для высококонтентного скрининга.
сайт www.perkinelmer.com

Рисунок 19в. Многообразие микроскопов в медицине. Бумажный микроскоп.
сайт www.mccrone.com
Флуоресцентные микроскопы за счет развития флуоресцентных проб и белков позволяют «рассматривать» биохимические процессы в реальном времени. Флуоресцентные белки помогают увидеть увеличение внутриклеточной концентрации кальция и магния, концентрации важных метаболитов (NADH+, лактата, глутамата и многих других), изменения pH, потенциалов действия, развитие важнейших клеточных процессов —прохождение клетки по клеточному циклу, программируемую клеточную гибель, активацию сигнальных каскадов и много другого.
Огромные возможности микроскопии, естественно, нашли применение в разработке новых лекарств, в том числе и для скрининга огромных библиотек химических веществ, антисмысловых РНК, антител и т.п. Микроскопия позволяет идентифицировать комплексные фенотипы, не ограничиваясь, как в биохимических скринингах, лишь одной мишенью. Микроскопы для высококонтентного анализа (high-content screening/analysis) потеряли свои канонические окуляры и открытые объективы — эти приборы выглядят как закрытые ящики, подключенные к компьютеру, однако могут снимать тысячи экспериментальных точек в день (особенно если они интегрированы с роботами для автоматической подачи образцов). Результаты таких анализов также не обрабатываются вручную, а анализируются компьютером по заранее заданным исследователем правилам. Компьютерный анализ применяется все шире, и, безусловно, скоро распространится и на рутинные научные исследования, и на постановку диагнозов. Последнее преимущество позволяет уменьшить субъективность диагноста: если раньше диагноз выставляли лишь с помощью взгляда патолога, то сейчас возможно применять программы для анализа, которые не будут зависеть от неопытности или ошибок врача. Оцифровка микроскопических изображений позволит анализировать их массивы с помощью машинного обучения, и в особо сложных случаях получать консультации врачей из любой точки мира.
Развитие биологии все время демонстрировало сложность и неоднородность устройства биологических объектов — от клетки к органу в целом. Микроскоп позволяет сохранить часть этой сложности, рассматривая клетки и ткани как комплексные системы, а не просто множество реакций в пробирке. И потому этому старейшему биологическому инструменту можно спокойно предсказать еще долгие годы жизни.
Календарь
На основе статей спецпроекта мы решили сделать календарь «12 методов биологии» на 2019 год. Эта статья представляет сентябрь.

Литература
- H. Gest. (2004). The discovery of microorganisms by Robert Hooke and Antoni van Leeuwenhoek, Fellows of The Royal Society. Notes and Records of the Royal Society. 58, 187-201;
- Giuseppe Musumeci. (2014). Past, present and future: overview on histology and histopathology. J Histol Histopathol. 1, 5;
- Abbe E. (1883). XV. The relation of aperture and power in the microscope. J. R. Microsc. Soc. 3, 790–812;
- Hannah Landecker. (2009). Seeing things: from microcinematography to live cell imaging. Nat Meth. 6, 707-709;
- Rusk N. (2009). The fluorescence microscope. Nat. Methods;
- Иммунитет: борьба с чужими и… своими;
- Моноклональные антитела;
- Osamu Shimomura, Frank H. Johnson, Yo Saiga. (1962). Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan,Aequorea. J. Cell. Comp. Physiol.. 59, 223-239;
- Флуоресцентные белки: разнообразнее, чем вы думали!;
- Флуоресцирующая Нобелевская премия по химии;
- Miyawaki A., Llopis J., Heim R., McCaffery J.M., Adams J.A., Ikura M. et al. (1997). Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 388, 882–887;
- Sheppard C.J.R. and Wilson T. (1981). The theory of the direct-view confocal microscope. J. Microsc. 124, 107–117;
- W Denk, J. Strickler, W. Webb. (1990). Two-photon laser scanning fluorescence microscopy. Science. 248, 73-76;
- По ту сторону дифракционного барьера: Нобелевская премия по химии 2014;
- Лучше один раз увидеть, или Микроскопия сверхвысокого разрешения;
- Stefan W. Hell, Jan Wichmann. (1994). Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett.. 19, 780;
- Флуоресцентные репортеры и их молекулярные репортажи;
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, et. al.. (2006). Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science. 313, 1642-1645.

Комментарии


