Мамонты, кости и лекарственная устойчивость: новые технологии позволяют изучать эволюцию возбудителей инфекционных заболеваний
31 октября 2013
Мамонты, кости и лекарственная устойчивость: новые технологии позволяют изучать эволюцию возбудителей инфекционных заболеваний
- 1040
- 0
- 0
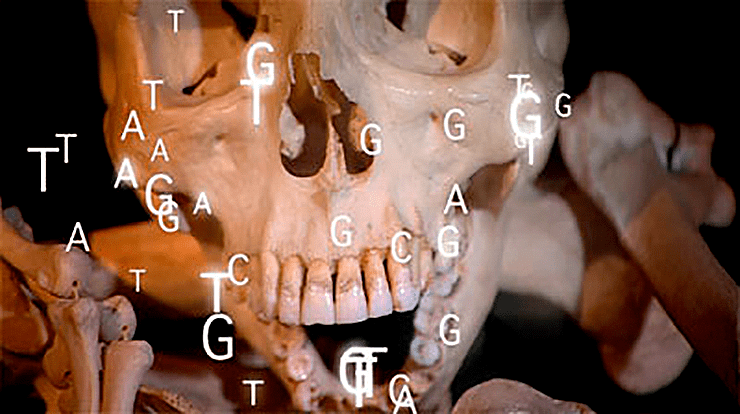
ДНК патогенных бактерий, извлеченная из археологических находок, помогает изучать эволюцию современных возбудителей инфекционных заболеваний
-
Автор
-
Редакторы
Статья на конкурс «био/мол/текст»: Изучение эволюции патогенных микроорганизмов до последнего времени сталкивалось с объективными трудностями, главной из которых было отсутствие ископаемых остатков их предковых форм. Как же узнать, какими были микроорганизмы, вызывавшие опустошительные эпидемии в древности и в средние века? Новые технологии, позволяющие изучать древнюю ДНК, обнаруживаемую в археологическом материале, а также современные методы эволюционного анализа, позволяют реконструировать историю древних эпидемий и искать предшественников современных возбудителей инфекционных заболеваний.

Конкурс «био/мол/текст»-2013
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2013 в номинации «Лучший обзор».
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific. Спонсор приза зрительских симпатий — фирма Helicon.
Изучение эволюции живых организмов невозможно представить без изучения их предков и «родственников» — отдаленных и близких. Долгое время считалось, что палеонтологические методы малоприменимы для изучения эволюции микроорганизмов, поскольку относительно малые размеры и отсутствие скелета не позволяют им сохраняться в окаменелостях в доступном для изучения виде. Исключение составляют, пожалуй, только цианобактерии, которые оказались способными формировать окаменелости двух типов: строматолиты (прижизненно минерализованные многослойные колонии) и микрофоссилии (одиночные минерализованные бактериальные клетки).
Значительный прогресс в палеомикробиологии был достигнут относительно недавно, когда было выяснено, что отдельные биологические молекулы могут сохраняться в палеонтологических образцах в течение миллионов лет. К счастью, одной из таких молекул является ДНК, что позволяет использовать обнаруженные фрагменты этого вещества для идентификации древних микроорганизмов и изучения их биологического разнообразия.
«Рекордсменами по выживанию» являются ДНК микроорганизмов, законсервированных внутри кусочков янтаря. В янтаре возрастом 25–35 млн. лет обнаружены, в частности, фрагменты ДНК стафилококков [1]. Применительно к микроорганизмам, вызывающим эпидемические инфекционные заболевания у человека, интерес представляют более поздние находки, связанные не только с палеонтологическим, но и с археологическим материалом. Как оказалось, возбудители инфекционных заболеваний, которые сопровождаются бактериемией (то есть проникновением возбудителя в кровь), — такие, например, как возбудители чумы, сыпного тифа и т.д., — заносятся с током крови во внутренние ткани зубов (пульпу). ДНК этих возбудителей сохраняется в пульпе зубов после смерти заболевшего, причем, благодаря особенностям анатомического строения зубов, проникновение посторонних микроорганизмов в пульпу практически не происходит.
Для того, чтобы извлечь бактериальную ДНК из пульпы зубов, предложено несколько остроумных способов. Один из них состоит в том, что зуб извлекают из черепа, обрабатывают его поверхность антисептиком, заливают слоем стерильной резины (рис. 1а) для того, чтобы избежать контаминации (загрязнения) чужеродной ДНК на последующих этапах работы, разрезают верхушку зуба в стерильных условиях, чтобы открыть доступ к пульпе (рис. 1б и в), впрыскивают туда стерильной иглой раствор, который растворяет ДНК (рис. 1г), а затем помещают зуб в стерильную пробирку (рис. 1д) и центрифугируют на высокой скорости (рис. 1е) для того, чтобы содержащий ДНК раствор под действием центробежной силы перешел в пробирку. Количество полученной ДНК можно затем многократно увеличить, используя полимеразную цепную реакцию (ПЦР) [12].

Рисунок 1. Способ экстракции ДНК из пульпы зуба.
В полимеразной цепной реакции используются уникальные одноцепочечные фрагменты ДНК (так называемые праймеры), которые позволяют распознать в изучаемой пробе и многократно увеличить количество (амплифицировать) только ДНК определенного вида микроорганизмов, — например, ДНК возбудителя чумы и никакую иную. Использование ПЦР для обнаружения микробной ДНК в ископаемых останках и археологическом материале позволило сделать множество совершенно удивительных открытий.
Так, например, установлено, что опустошительную эпидемию «Черной смерти», разразившуюся в Европе в середине XIV столетия, вызвали два штамма возбудителя чумы, которые были занесены независимо друг от друга и распространялись разными путями — первый с юга Европы на север, а второй — по побережью Балтийского и Северного морей, вызывая вспышки этой смертельной инфекции в ганзейских городах [3], [4].
ПЦР помогает также реконструировать исторические события: например, с использованием этого метода установлено, что осада города Дуэ французами во время войны за испанский престол (1710–1712 гг.) сопровождалась вспышкой сыпного тифа, возникшей, вероятно, вследствие заноса возбудителя этого заболевания — риккетсии — в Европу испанскими солдатами из Нового Света [5]. ДНК риккетсий обнаруживалась в пульпе зубов испанских солдат, массовые захоронения которых найдены в окрестностях этого города.
Впечатляющие результаты достигнуты благодаря палеомикробиологическим исследованиям в области изучения возбудителя туберкулёза. Турбекулез зачастую осложняется гематогенной диссеминацией возбудителя (т.е. распространением его через кровь) и поражением костей, — в частности, костей позвоночника. ДНК микобактерий туберкулёза может быть извлечена из пораженной костной ткани и использована для исследований. Кроме того, некоторые липидные факторы вирулентности микобактерий могут неплохо сохраняться в мумифицированных останках животных и человека, что позволяет подтверждать диагноз туберкулёза.
Выяснено, что туберкулезом страдали животные, на которых охотился первобытный человек — бизоны и мамонты [6]. Кроме того, очевидно, что туберкулёзом были поражены и первобытные люди, начиная с железного века. Возбудитель туберкулеза был обнаружен в костях людей из захоронения Аймырлыг на юге Сибири, датированного 2000 лет до н.э. [7], а также в костях детей из неолитической стоянки Атлит на территории современного Израиля, возраст которой определен в 9000 лет [8]. Учитывая, что поселение Атлит является самым древним из известных центров одомашнивания диких животных, не исключено, что первобытные люди заражались туберкулёзом от одомашненного крупного рогатого скота. ДНК микобактерий туберкулёза с большой частотой определялась в костях жителей Древнего Египта, начиная с 3500 лет до н.э.
Таким образом, туберкулез являлся частой инфекцией в древности. Однако ученых интересует вопрос о том, вызывали ли древние эпидемии те же варианты возбудителя, что и сейчас, или это были совершенно другие штаммы.
Применительно к возбудителю туберкулёза наибольший интерес вызывает происхождение штаммов генетического семейства Beijing (произносится как «Бэйджинг»), впервые выявленного в гистологических препаратах лёгочной ткани 1956–1990 гг. от больных из предместий Пекина. Штаммы этого генетического семейства являются наиболее контагиозными (т.е. заразными) и легче всего приобретают устойчивость к противотуберулезным препаратам, что делает весьма затруднительным лечение и ухудшает прогноз заболевания. Ответить на вопрос о происхождении штаммов Beijing помогают филогенетические исследования, основанные на анализе ДНК современных микобактерий туберкулеза, распространенных в различных географических регионах Земли в различных этнических группах.
Для изучения популяционной структуры микобактерий туберкулёза используются различные молекулярно-генетические методы. Одним из наиболее распространенных является метод MIRU—VNTR, основанный на обнаружении в ДНК микроорганизма специфических повторяющихся последовательностей — тандемных повторов. Родственные культуры возбудителя имеют совпадающее число тандемных повторов в специальных участках хромосомы (называемых MIRU—VNTR-локусами). На основании MIRU—VNTR метода можно изучить эволюционные отношения между отдельными штаммами, входящими в генетическое семейство Beijing и построить филогенетическое дерево, отражающее эволюционные связи между этими штаммами (подобное неукорененное дерево приведено на рис. 5).

Рисунок 5. Филогенетическое дерево 48 основных штаммов микобактерий туберкулёза внутри генотипа Beijing M. tuberculosis, представляющих различные географические регионы. Каждая ветвь представляет одно генетическое событие (одно изменение в одном локусе ДНК). Пунктирные линии обозначают возможные альтернативы.
Нетрудно заметить, что наиболее распространенным штаммом (генотипом) является генотип M11, который располагается в центре филогенетического дерева. Это, по-видимому, означает, что данный генотип является наиболее древним — родоначальником других штаммов. Если оценить современное географическое распространение этого штамма в Евразии, то окажется, что наиболее широко он распространен в Китае; при этом его доля убывает в направлении с востока на запад до северо-запада Российской Федерации. В то же время, в странах Западной Европы генотип M11 практически не встречается (рис. 6).

Рисунок 6. Географическое распределение основных MIRU-типов генотипа Beijing M. tuberculosis
Размер диаграмм на рис. 6 приблизительно пропорционален доле штаммов Beijing в общей популяции M. tuberculosis. Цифрами обозначен индекс генетического разнообразия (чем ближе этот показатель к единице, тем более разнообразна популяция возбудителя туберкулёза) [9]. Данные, представленные на рисунках 5 и 6, позволяют предположить, что генетическое семейство Beijing сформировалось в Китае (поскольку наиболее древняя его ветвь преобладает именно в Северном Китае), а затем распространялось в западном направлении, что и обеспечило «градиент» частоты его встречаемости. Наиболее вероятной гипотезой, объясняющей распространение возбудителя туберкулёза в направлении с Востока на Запад, является миграция населения, произошедшая в историческом прошлом.
Но какая же миграция населения могла привести к распространению этого генетического семейства туберкулеза в Евразии? Поскольку Beijing не является эндемичным для Европы, можно исключить миграции, которые в равной степени затрагивали Россию и Европу, как источник исключительного проникновения штаммов Beijing в Россию. К таковым относятся финно-угорская, скифская, и гуннская экспансии [10]. Выдвинуто предположение [9], что проникновение штаммов Beijing в Россию могло быть связано с евразийской экспансией монгольской империи Чингисхана в 13–15 веках. Средневековая монгольская империя впервые в истории тесно связала отдаленные части евразийского суперконтинента и создала единую экономическую и культурную систему, охватывавшую огромное пространство Евразии. Связи монголо-татар с Русью были тесными и продолжительными (в отличие от контактов с европейцами), что, вероятно, и обеспечило укоренение на территории России штаммов генетического семейства Beijing.
Таким образом, изучение эволюции патогенных микроорганизмов во многом связано с изучением истории человечества, эпидемий, войн и вызванных ими миграций. Междисциплинарные исследования, использующие данные естественных и гуманитарных наук, позволяют глубже понять картину совместного сосуществования популяций микроорганизмов и человеческого общества.
Литература
- L. H. Lambert, T. Cox, K. Mitchell, R. A. Rossello-Mora, C. Del Cueto, et. al.. (1998). Staphylococcus succinus sp. nov., isolated from Dominican amber. International Journal of Systematic Bacteriology. 48, 511-518;
- Lam Tran-Hung, Ny Tran-Thi, Gérard Aboudharam, Didier Raoult, Michel Drancourt. (2007). A New Method to Extract Dental Pulp DNA: Application to Universal Detection of Bacteria. PLoS ONE. 2, e1062;
- Stephanie Haensch, Raffaella Bianucci, Michel Signoli, Minoarisoa Rajerison, Michael Schultz, et. al.. (2010). Distinct Clones of Yersinia pestis Caused the Black Death. PLoS Pathog. 6, e1001134;
- Это чума;
- Tung Nguyen-Hieu, Gérard Aboudharam, Michel Signoli, Catherine Rigeade, Michel Drancourt, Didier Raoult. (2010). Evidence of a Louse-Borne Outbreak Involving Typhus in Douai, 1710-1712 during the War of Spanish Succession. PLoS ONE. 5, e15405;
- Oona Y-C. Lee, Houdini H. T. Wu, Helen D. Donoghue, Mark Spigelman, Charles L. Greenblatt, et. al.. (2012). Mycobacterium tuberculosis Complex Lipid Virulence Factors Preserved in the 17,000-Year-Old Skeleton of an Extinct Bison, Bison antiquus. PLoS ONE. 7, e41923;
- G. M. Taylor, E. Murphy, R. Hopkins, P. Rutland, Y. Chistov. (2007). First report of Mycobacterium bovis DNA in human remains from the Iron Age. Microbiology. 153, 1243-1249;
- Israel Hershkovitz, Helen D. Donoghue, David E. Minnikin, Gurdyal S. Besra, Oona Y-C. Lee, et. al.. (2008). Detection and Molecular Characterization of 9000-Year-Old Mycobacterium tuberculosis from a Neolithic Settlement in the Eastern Mediterranean. PLoS ONE. 3, e3426;
- Мокроусов И.В. Генетическое разнообразие и эволюция Mycobacterium tuberculosis: автореф. дис. ... док. мед. наук. — СПб., 2009;
- P. A. UNDERHILL, G. PASSARINO, A. A. LIN, P. SHEN, M. MIRAZON LAHR, et. al.. (2001). The phylogeography of Y chromosome binary haplotypes and the origins of modern human populations. Ann Human Genet. 65, 43-62;
- Сверим часы;
- 12 методов в картинках: полимеразная цепная реакция.

Комментарии


